Clear Sky Science · nl
Een end-to-end raamwerk voor reactiviteit in heterogene katalyse
Waarom snellere katalysatorontwerp ertoe doet
De moderne samenleving steunt op katalysatoren voor de productie van brandstoffen, kunststoffen, meststoffen en talloze alledaagse producten. Toch is het vinden van betere katalysatoren vaak als zoeken naar een speld in een hooiberg, omdat elk materiaal tegelijkertijd duizenden mogelijke microscopische reacties kan bevorderen. Dit artikel introduceert CARE, een nieuw computationeel raamwerk dat slimme regels en machine learning gebruikt om deze verwarde reactienetten veel sneller en vollediger in kaart te brengen en te simuleren dan voorheen. Daarmee belooft het schonere energietechnologieën en efficiëntere chemische processen te sturen, terwijl de rekencapaciteit sterk vermindert.
Het ontwarren van drukke reactiepaden
Op het oppervlak van een vaste katalysator volgen binnenkomende moleculen niet simpelweg één keurig pad van reaktant naar product. In plaats daarvan bewegen ze zich door een doolhof van kortlevende intermediaten en concurrerende routes. Traditionele computermethoden vertrouwen op menselijke intuïtie om een beperkte set mogelijke stappen te kiezen en gebruiken vervolgens kwantitatieve berekeningen om hun energieën te evalueren. Dat werkt voor kleine netwerken maar faalt snel naarmate systemen complexer worden, waarbij zeldzame routes die de langetermijnactiviteit, deactivering of selectiviteit bepalen over het hoofd worden gezien. CARE pakt deze uitdaging aan door automatisch zeer grote reactienetwerken op te bouwen uit eenvoudige bouwregels, waarbij gegarandeerd alle plausibele bindingsbrekende en bindingsvormende gebeurtenissen tussen koolstof, waterstof en zuurstof worden opgenomen, ook die chemici normaal gesproken zouden uitsluiten.
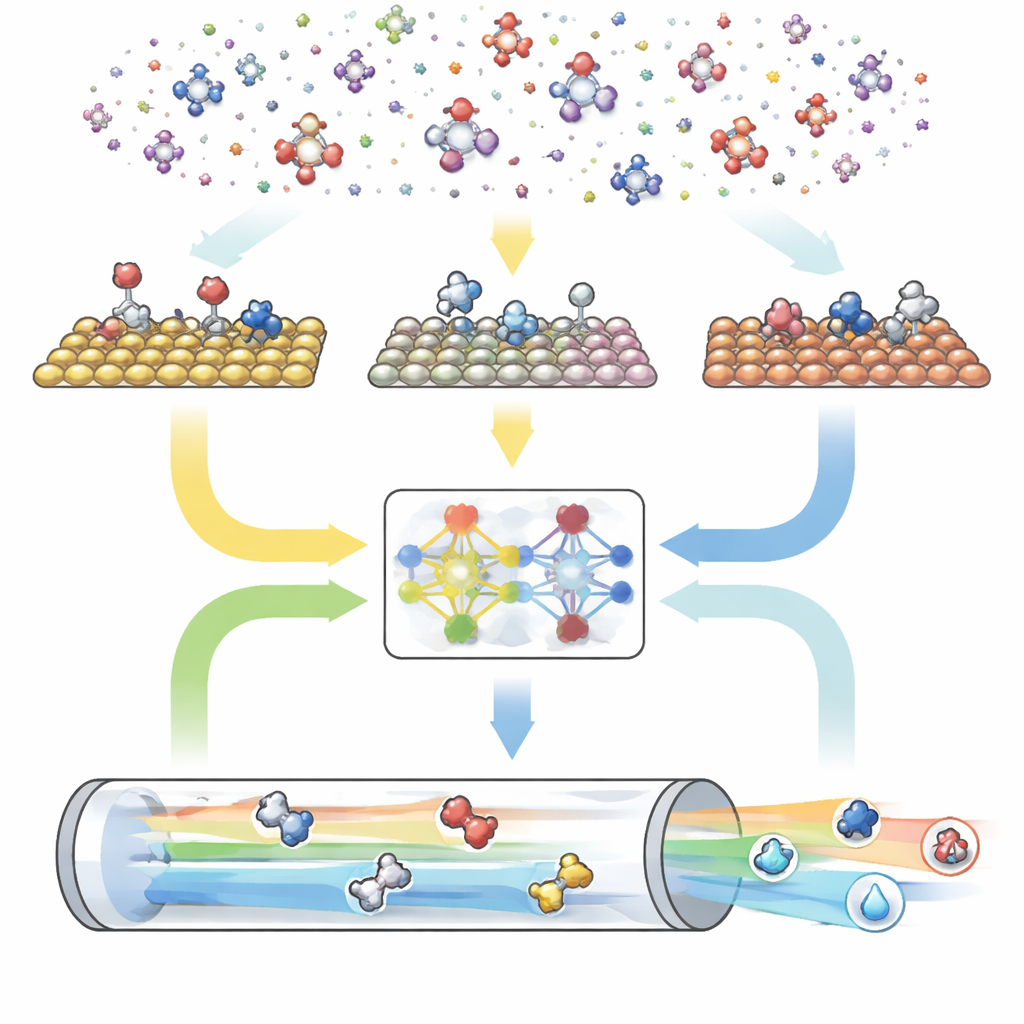
Een drietalige digitale motor voor reacties
CARE is opgebouwd als een end-to-end pijplijn met drie hoofdmodules. Ten eerste definieert een regelgebaseerde generator de “chemische ruimte” door het maximale aantal koolstof- en zuurstofatomen te kiezen en vervolgens eenvoudige sjablonen toe te passen om alle overeenkomende moleculen en hun oppervlaktegebonden vormen te creëren. Ten tweede roept een energie-evaluatiemodule moderne machine-learningmodellen aan—vooral een graafneurale netwerkaanpak genaamd GAME-Net-UQ—om de energieën van intermediaten en overgangstoestanden op vele metaaloppervlakken te schatten. Dit model behandelt elke structuur als een netwerk van atomen en bindingen, geeft zowel een energie als een onzekerheid terug, en is nauwkeurig binnen enkele tienden van een elektronvolt terwijl het lichtgewicht en snel blijft. Ten derde gebruikt een microkinetische solver deze energieën om te berekenen hoe alle reacties gezamenlijk verlopen onder realistische condities van temperatuur, druk, spanning en pH, en voorspelt zo totale reactiesnelheden, oppervlaktesaturaties en productselectiviteit.
Praktijktests: brandstofmoleculen en klimaatchemie
Om aan te tonen dat CARE niet slechts een theoretische oefening is, passen de auteurs het toe op drie industrieel relevante vraagstukken van toenemende moeilijkheidsgraad. Voor methanoldecompositie—een reactie die belangrijk is voor waterstofopslag—genereren ze een bescheiden netwerk en evalueren het voor vele metaal-katalysatoren en kristalvlakken. CARE reproduceert de bekende “vulkaan”-trend in activiteit en identificeert correct ruthenium als een van de beste presteerders, in overeenstemming met experimenten, maar met een fractie van de rekentijd die volledige kwantumberekeningen vergen. Vervolgens richten ze zich op de elektrochemische omzetting van kooldioxide op koper, met de nadruk op hoe drie-koolstofproducten zoals 1-propanol en propeen ontstaan. Door speciale stappen op te nemen die rekening houden met protonen, elektronen en oplossingscondities, vangt CARE hoe pH en aangelegde spanning paden verschuiven en voorspelt het correct dat 1-propanol wordt bevoordeeld boven propeen, in overeenstemming met eerdere gedetailleerde studies.
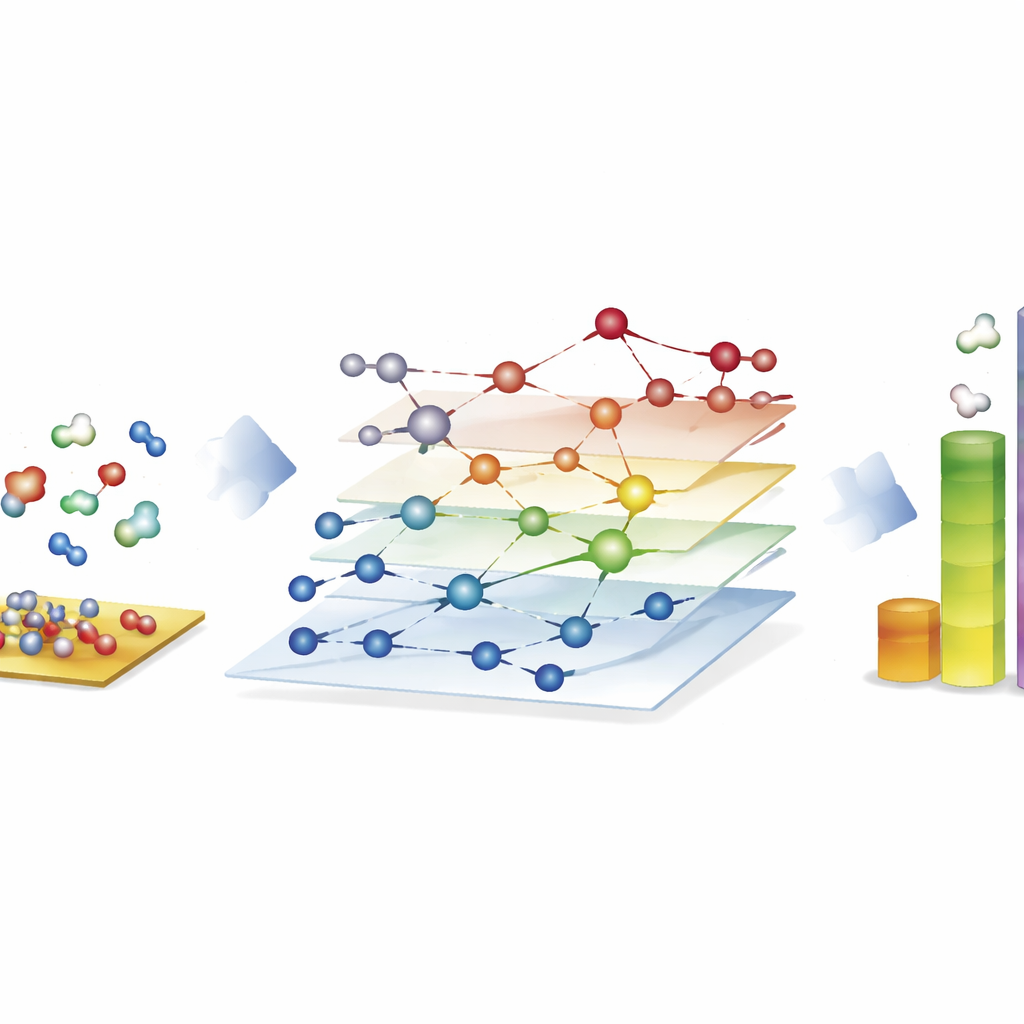
Het verkennen van enorme reactienetten voor synthetische brandstoffen
De meest opvallende demonstratie komt van het Fischer–Tropsch-proces, dat mengsels van koolmonoxide en waterstof omzet in lange-keten koolwaterstoffen voor brandstoffen en chemicaliën. Hier bouwen de auteurs netwerken met bijna 40.000 oppervlakte-soorten en ongeveer 370.000 elementaire reacties—ver buiten wat traditionele, op kwantum gebaseerde studies volledig kunnen onderzoeken. Met CARE evalueren ze alle intermediaten en belangrijke reactiedrempels op kobalt-, ijzer-, nikkel- en rutheniumoppervlakken in slechts enkele uren op standaardhardware, een snelheidswinst van ongeveer een miljoen keer vergeleken met directe kwantumberekeningen. Microkinetische simulaties op deze netwerken reproduceren bekende trends: kobalt en ijzer vormen bij voorkeur langere koolwaterstofketens, ijzer produceert meer kooldioxide via nevenreacties, en nikkel neigt naar krachtigere hydrogenatie. Hoewel sommige details, zoals methaanopbrengsten, onvolmaakt blijven, laat het raamwerk zien welke bindingsvormende stappen de ketengroei domineren en benadrukt waar modellen nog verfijning nodig hebben.
Wat dit betekent voor toekomstige katalysatoren
Voor niet-specialisten is de kernboodschap dat CARE een praktische manier biedt om enorme reactieruimtes op katalytische oppervlakken te verkennen die voorheen buiten bereik lagen. Door netwerkgeneratie te automatiseren, snelle machine-learning “surrogaat”-modellen voor kwantumenergieën in te schakelen en de resulterende kinetiek efficiënt op te lossen, kan het kandidaat-katalysatoren rangschikken, veelbelovende bedrijfscondities identificeren en onverwachte paden blootleggen met veel minder menselijke vooringenomenheid en rekenkundige kosten. Hoewel de auteurs resterende uitdagingen noemen—zoals een betere behandeling van drukbezette oppervlakken, solvenseffecten en nog grotere netwerken—wijst het werk op een toekomst waarin computers complexe reacties snel kunnen screenen, van kooldioxide-reductie tot plasticrecycling en biomassaupgrading, en experimenten sturen naar de meest veelbelovende ideeën in plaats van ontdekking aan toeval over te laten.
Bronvermelding: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Trefwoorden: heterogene katalyse, reactienetwerken, machine learning, microkinetische modellering, Fischer–Tropsch-synthese