Clear Sky Science · nl
Ongekende robuustheid van fysica-geïnformeerde atomaire energiemodellen bij en boven kamertemperatuur
Waarom dit belangrijk is voor alledaagse chemie
Computersimulaties zijn de werkpaarden van moderne chemie en materialenwetenschap. Ze laten onderzoekers moleculen in silico draaien, vibreren en botsen observeren in plaats van in dure, tijdrovende experimenten. Maar wanneer deze simulaties op machinaal leren vertrouwen, kunnen ze plotseling "exploderen" en onmogelijke molecuulvormen produceren—vooral bij hogere temperaturen. Deze studie introduceert een nieuw type fysica-geïnformeerd machinaal-lerend model dat zulk onderzoek voor extreem lange tijden kan uitvoeren, bij temperaturen tot 1000 kelvin, zonder in te storten.
Van slimme snelkoppelingen naar fragiele simulaties
Traditionele kwantumchemie berekent moleculaire energieën met hoge nauwkeurigheid maar is pijnlijk traag. Eenvoudigere krachvelden zijn snel maar vaak benaderend. Machinaal-geleerde potentialen proberen het beste van beide werelden te combineren: ze leren een snelkoppeling van moleculaire geometrie naar energie en krachten, en gebruiken die vervolgens om moleculaire dynamica aan te sturen. Op papier lijken veel van zulke modellen uitstekend, met zeer kleine gemiddelde fouten op standaard testsets. In de praktijk kunnen die cijfers misleidend zijn. Wanneer moleculen tijdens een simulatie nieuwe vormen verkennen—vooral bij hogere temperaturen—worden veel modellen buiten het bereik van de structuren waarop ze getraind zijn geduwd. In plaats van moleculen zachtjes terug te sturen naar realistische vormen, kunnen ze krachten voorspellen die bindingen uitrekken of samendrukken totdat het hele systeem onfysisch wordt en de simulatie crasht.
Modellen bouwen op quantum-gebaseerde bouwstenen
De auteurs pakken deze fragiliteit aan door te veranderen wat het model leert en hoe het door voorafgaande fysica wordt gestuurd. Ze gebruiken een raamwerk genaamd FFLUX, dat voortbouwt op de Interacting Quantum Atoms (IQA)-benadering. In IQA wordt een molecule verdeeld in "topologische atomen" waarvan de individuele energieën rechtstreeks uit de kwantummechanica worden bepaald. Deze atomaire energieën zijn fysisch betekenisvol en tellen op tot de totale energie van de molecule. In plaats van willekeurige site-energieën te leren, leren de nieuwe Gaussian process-modellen deze in de quantumwortel gewortelde atomaire energieën, wat een diep fysisch anker voor elke voorspelling biedt. Vier flexibele organische moleculen—peptide-afgesloten glycine en serine, malondialdehyde en aspirine—dienen als uitdagende testgevallen vanwege hun vele interne bewegingen en de bekende moeilijkheid voor bestaande machinaal-geleerde krachvelden.
Het model leren problemen te verwachten
Een belangrijke innovatie ligt in hoe de Gaussian process wordt opgezet voordat hij ooit data ziet: zijn "mean function", die encodeert wat het model veronderstelt in slecht bekende gebieden. Het merendeel van eerder werk stelt deze mean simpelweg op nul, en doet alsof het model geen voorafgaande verwachtingen heeft. De auteurs verschuiven deze mean daarentegen bewust richting hogere atomaire energietoestanden, terwijl ze het fysisch zinnig houden. Deze ontwerpkeuze betekent dat wanneer het model moet extrapoleren—bijvoorbeeld wanneer bindingen tijdelijk overstrekt zijn—het van nature de voorkeur geeft aan voorspellingen die extreme vervormingen bestraffen. In uitgebreide tests gedroegen versies van het model die alleen verschilden in deze prior zich zeer verschillend. Modellen met conventionele of laag-energetische means overleefden vaak minder dan een picoseconde voordat een molecule explodeerde of implodeerde. Daarentegen leverde de beste hoge-energie mean (gedoopt MF5) simulaties op die gedurende het volledige nanoseconde-testvenster stabiel bleven bij temperaturen van 300 tot 1000 kelvin voor alle vier de moleculen.
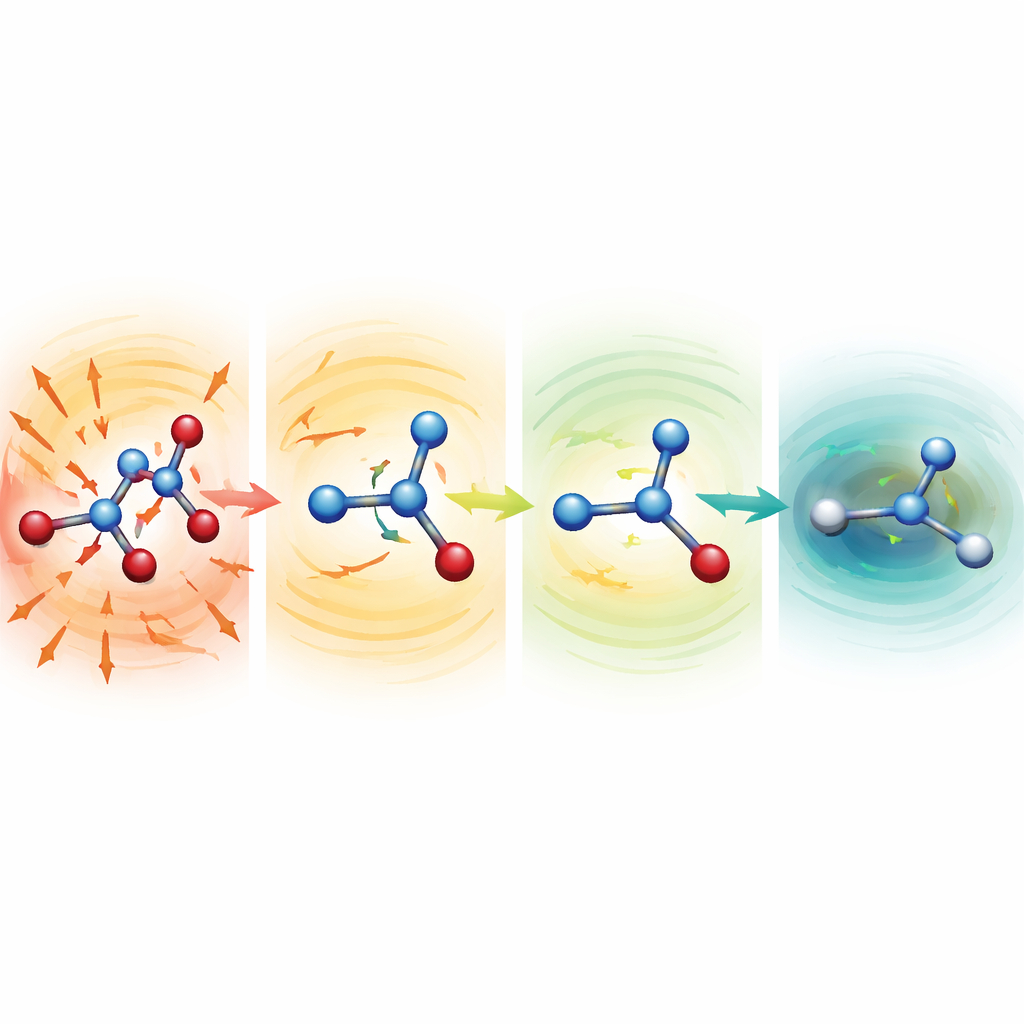
Zien hoe vervormde moleculen zichzelf genezen
Om te onderzoeken waarom de robuuste modellen zo goed werken, startten de onderzoekers simulaties vanaf opzettelijk verminkte structuren met ernstig uitgerekte of samengeperste bindingen. Voor serine, aspirine en malondialdehyde lagen deze beginsituaties honderden tot meer dan duizend kilocalorieën per mol boven een normale structuur—configuraties die normaal verwoestend zouden zijn. Met zwakkere mean-functies vielen de moleculen snel uiteen. Met de MF5-opzet wezen de voorspelde krachten echter direct in de richting van het verkorten van uitgerekte bindingen en het verlengen van samengedrukte. Binnen tientallen tot honderden tijdstappen ontspanden de moleculen tot realistische vormen en bleven daarna stabiel evolueren. Het team toonde ook aan dat, zonder ooit te zijn getraind op krachten, dezelfde modellen geometrie-optimalisaties van alanine-dipeptide kunnen sturen, waarbij bekende laag-energetische conformaties en relatieve energieën binnen een paar tienden van een kilocalorie per mol worden gereproduceerd, maar ongeveer 200 keer goedkoper dan volledige kwantumberekeningen.
Lange, hete simulaties op alledaagse hardware
Robuustheid gaat niet alleen over het overleven van één moeilijke start; het gaat erom stand te houden over miljoenen of miljarden tijdstappen. De auteurs daagden hun beste modellen verder uit door 50 onafhankelijke simulaties bij 500 kelvin te draaien, elk van 10 nanoseconden, voor hun vier testmoleculen. Geen van deze runs crashte, wat een gecombineerde simulatieperiode van een halve microseconde opleverde—ongewoon voor geavanceerde machinaal-geleerde krachvelden. Nog opvallender: de simulaties draaiden efficiënt op standaard CPU's, stap-voor-stap concurrerend met of beter dan sommige prominente neurale-netwerk potentialen die krachtige GPU's vereisen. Gedurende de simulaties verkenden de moleculen rijke verzamelingen vormen en metastabiele toestanden, wat aantoonde dat robuustheid niet werd bereikt door beweging kunstmatig te bevriezen of starre structuren af te dwingen.
Wat dit betekent voor toekomstige moleculaire modellering
Voor niet-experts is de kernboodschap dat niet alle laag-fout machinaal-geleerde modellen betrouwbaar zijn wanneer ze zwaar worden belast. Door hun modellen te verankeren in quantum-afgeleide atomaire energieën en de ingebouwde verwachtingen van het model zorgvuldig af te stemmen op hoge-energetische toestanden, creëerden de auteurs een familie potentialen die van nature "herstellende krachten" produceren—het moleculaire equivalent van een veiligheidstuigje—dat simulaties fysisch houdt, zelfs bij hoge temperaturen en vanuit vervormde beginsituaties. Deze benadering belooft betrouwbaardere en langere simulaties van complexe moleculen, en wijst naar toekomstige uitbreidingen waarin soortgelijke fysica-geïnformeerde modellen gecondenseerde fasen en subtiele interacties zoals dispersie kunnen behandelen, terwijl ze praktisch blijven qua rekenkosten.
Bronvermelding: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Trefwoorden: machinaal leren krachvelden, robuustheid moleculaire dynamica, Gaussian process potentialen, fysica-geïnformeerde modellering, quantum atomaire energieën