Clear Sky Science · nl
haCCA: multi-module Integratie van spot-gebaseerde ruimtelijke transcriptomen en metabolomen
Waarom het belangrijk is moleculen op hun plek in kaart te brengen
Onze lichamen bestaan uit talloze kleine celbuurten, elk met een eigen samenstelling van actieve genen en chemische stoffen. Tot voor kort moesten onderzoekers deze moleculen bestuderen nadat weefsel was vermalen tot een homogene massa, waarbij elk gevoel voor "waar" verloren ging. Dit artikel introduceert een nieuwe computationele methode, haCCA, die twee krachtige beeldvormingstechnieken samenbrengt zodat onderzoekers ter plaatse kunnen zien hoe genen en kleine moleculen zijn gerangschikt in echte weefsels en tumoren. Zo’n kaart kan verborgen ziektepatronen onthullen en wijzen op meer gerichte behandelingen.
Twee verschillende blikvelden op hetzelfde weefsel
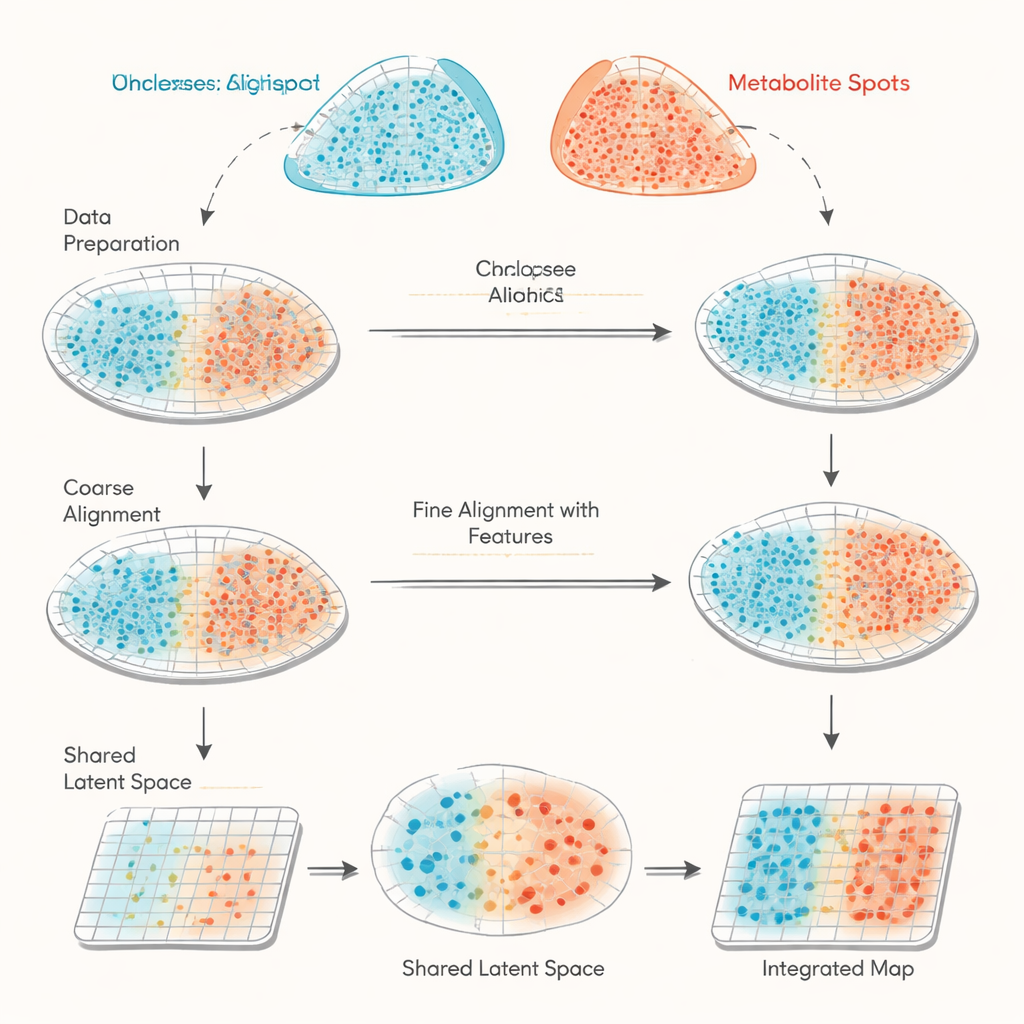
De studie richt zich op het combineren van gegevens uit twee ruimtelijke methoden die steeds vaker in de biologie worden gebruikt. Ruimtelijke transcriptomics registreert welke genen op honderden tot duizenden kleine spots in een weefselsnede actief zijn. MALDI massaspectrometrie-imaging meet de hoeveelheden van vele kleine moleculen, zoals metabolieten en lipiden, op vergelijkbaar dichte rasterpunten. Het probleem is dat deze twee instrumenten niet precies dezelfde posities meten of dezelfde set kenmerken hebben, waardoor hun data op twee niet-uitgelijnde kaarten met verschillende legenda’s lijken. Bestaande benaderingen proberen meestal alleen de vormen van weefselsecties op basis van coördinaten te matchen, wat onnauwkeurig kan zijn en geen goede manier biedt om te controleren hoe goed de uitlijning werkelijk is.
Een slimmer manier om moleculaire kaarten op één lijn te brengen
haCCA (afkorting van hierarchical anchor-guided canonical correlation analysis) pakt deze uitdaging aan door geometrie en biologie te combineren. Eerst voert het een tweestaps “morfologische uitlijning” uit van de spotrasters van de twee technologieën. Deskundigen selecteren enkele overeenkomende herkenningspunten op de weefselbeelden om grofweg verschuivingen en rotaties te corrigeren, waarna een geautomatiseerde stap afwijkingen fijn afstemt rond gescheurde randen of ontbrekende stukken. Vervolgens zoekt de methode naar "anker"-paren van spots die dicht bij elkaar liggen en zich in lokaal uniforme gebieden bevinden, waardoor ze waarschijnlijk hetzelfde weefselgebied representeren. Vanuit deze ankers berekent haCCA welke genen en metabolieten de neiging hebben samen te veranderen en brengt die terug tot een gedeelde laag-dimensionale representatie die hun sterkste gezamenlijke patronen vastlegt.
Correlaties omzetten in een eenduidig weefselbeeld
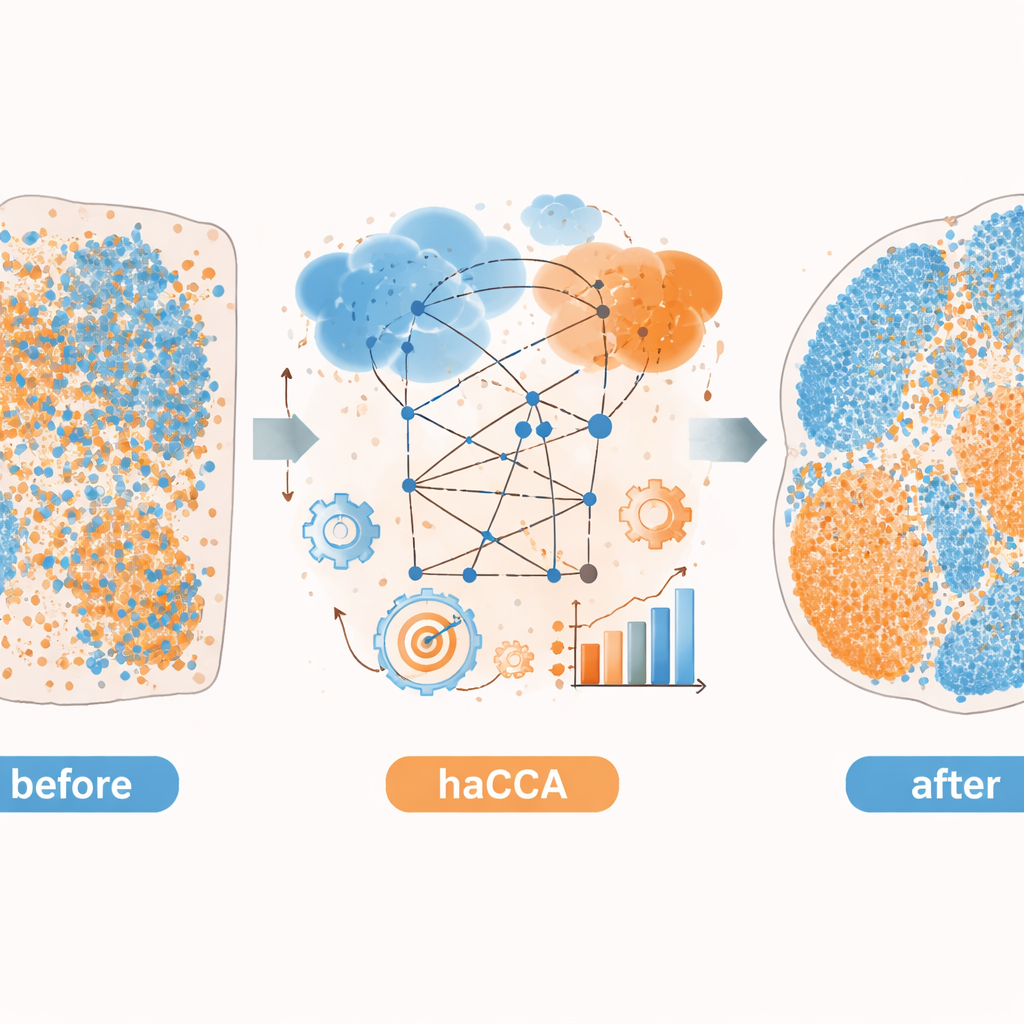
Met zowel ruimtelijke coördinaten als de gedeelde moleculaire representatie bepaalt haCCA via een optimaliseringsprobleem hoe waarschijnlijk het is dat elk gen-spot gekoppeld moet worden aan elk metaboliet-spot. Deze stap is ontworpen om spots zowel ruimtelijk dicht bij elkaar te houden als vergelijkbaar in hun gecombineerde gen–metabolietprofiel. Het eindresultaat is een "transportplan" dat elk punt in de ene dataset linkt aan zijn beste partner in de andere, waarmee een geïntegreerde multimodale kaart ontstaat. Op zorgvuldig geconstrueerde testgegevens—waarbij de ware relaties bekend zijn—tonen de auteurs aan dat elke fase van de workflow (grove uitlijning, verfijnde uitlijning en kenmerk-bewuste matching) de drie onafhankelijke nauwkeurigheidsmaatregelen geleidelijk verbetert. Vergeleken met andere tools die vooral op geometrie vertrouwen, behaalt haCCA consequent betere uitlijning en trouwere overdracht van regiolabels.
Verborgen biologie onthullen in hersen- en leverkanker
De auteurs passen haCCA vervolgens toe op echte muizenhersen- en levertumoren. Voor de hersenen integreren ze commerciële ruimtelijke transcriptomicsgegevens met metabolietbeelden van dezelfde of aangrenzende secties. De methode behoudt bekende metabolische territoria en reconstrueert verwachte overlap, zoals de co-locatie van dopamine met het gen dat het sleutelenzym codeert. Door genen en metabolieten gezamenlijk te clusteren, vinden ze dat de gecombineerde data genuanceerdere weefselsubregio’s onderscheiden dan één van beide modaliteiten afzonderlijk. In een preklinisch model van intrahepatische cholangiocarcinoom, een type leverkanker, gebruiken ze haCCA om tumoren te vergelijken die wel of niet neutrofiele extracellulaire vallen (webachtige structuren die door immuuncellen worden vrijgegeven) kunnen vormen. De geïntegreerde kaarten tonen aan dat, wanneer deze vallen aanwezig zijn, een gen genaamd Scd1 en de daaraan gerelateerde vetzuren verrijkt zijn in maligne regio’s, wat wijst op een verschuiving naar veranderd vetmetabolisme in de tumor.
Wat dit betekent voor toekomstig onderzoek
In gewone bewoordingen is haCCA te vergelijken met het uitlijnen van luchtfoto’s die met verschillende camera’s zijn genomen—de ene gevoelig voor gebouwcontouren, de andere voor warmtehandtekeningen—om een scherper beeld te krijgen van wat er in elk stadsblok gebeurt. Door nauwkeurig te combineren waar genen actief zijn met waar belangrijke metabolieten zich ophopen, helpt deze workflow wetenschappers om beide kanten van cellair gedrag tegelijk te profileren: instructies en de resulterende chemie. De aanpak verbetert eerdere uitlijningsmethoden, is verpakt in een toegankelijke Python-tool en kan worden uitgebreid naar andere ruimtelijke technologieën. Naarmate zulke geïntegreerde kaarten gebruikelijker worden, kunnen ze ons begrip verdiepen van hoe tumoren en andere weefsels hun metabolisme organiseren, reageren op behandeling en in de loop van de tijd evolueren.
Bronvermelding: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Trefwoorden: ruimtelijke multi-omics, transcriptomics, metabolomics, tumormetabolisme, gegevensintegratie