Clear Sky Science · nl
Ab initio-bepaling van fasestabiliteiten van dynamisch gedesoriënteerde vaste stoffen: rotatie C2-disorde in Li2C2
Waarom dit verschuivende vaste stof belangrijk is
Veel moderne technologieën vertrouwen op vaste stoffen die hun interne structuur stilletjes kunnen veranderen wanneer ze verwarmd of samengedrukt worden. Deze veranderingen, faseovergangen genoemd, zijn centraal voor toepassingen zoals vastestofkoeling en veiligere batterijen. Deze studie bekijkt een eenvoudig amalgaam, lithiumcarbide (Li2C2), dat overschakelt van een keurig geordende vorm naar een meer rusteloze, dynamisch gedesoriënteerde vorm naarmate de temperatuur stijgt. Door deze transformatie atoom voor atoom te volgen in computersimulaties laten de auteurs zien hoe de interne "rusteloosheid" van kleine moleculaire eenheden de balans tussen twee kristalstructuren kan doen kantelen.
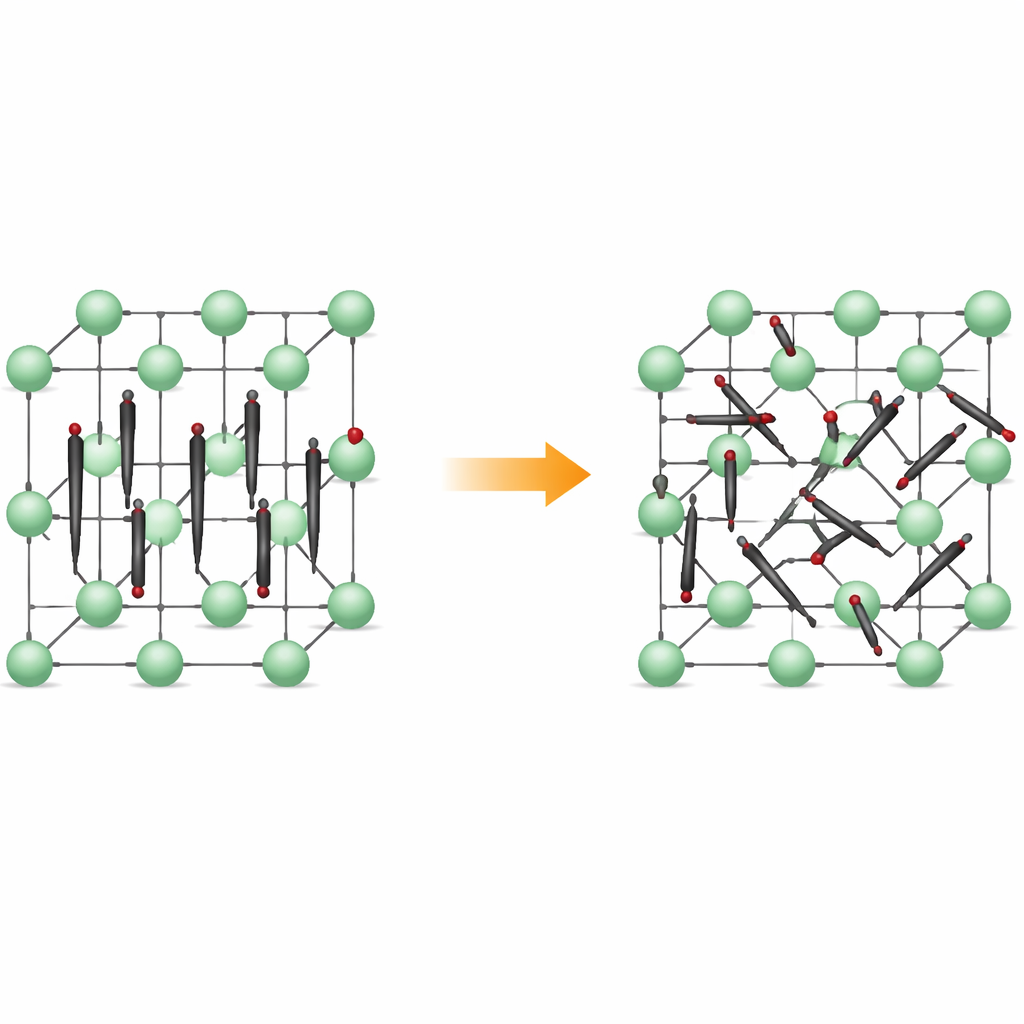
Van nette rijen naar rusteloze beweging
Bij lage temperaturen vormt Li2C2 een orthorombisch kristal: de koolstofatomen koppelen zich tot kleine C2-dimeren die vrijwel allemaal in dezelfde richting wijzen, als uitgelijnde lucifers. Lithiumionen bevinden zich daartussen en creëren een regelmatig, driedimensionaal raamwerk. Wanneer het materiaal wordt verwarmd transformeert het naar een kubische vorm, waarbij de posities van de dimercentra op een rooster geordend blijven, maar de dimeren zelf geen vaste oriëntatie meer aanhouden. In plaats daarvan roteren ze tussen meerdere voorkeuroriëntaties en verblijven ze in ondiepe energiedalletjes die overeenkomen met specifieke uitlijningen. Het materiaal blijft een vaste stof, maar zijn interne structuur wordt dynamisch gedesoriënteerd.
De verandering volgen langs een vloeiend pad
Om te begrijpen welke fase bij een gegeven temperatuur stabieler is, moet men hun vrije energieën vergelijken, die energie en entropie (een maat voor wanorde) combineren. Standaardmethoden gebaseerd op kleine vibraties rond vaste posities hebben moeite wanneer atomen aanzienlijk ronddolen of draaien. Hier gebruiken de auteurs een techniek genaamd stress–strain thermodynamische integratie, gebaseerd op eersteklas (ab initio) moleculaire dynamica. Ze construeren een vloeiend deformatiepád dat de simulatiecel continu vervormt van de orthorombische structuur bij lage temperatuur naar de kubische bij hoge temperatuur. Langs dit pad voeren ze lange simulaties uit bij vaste temperaturen en meten hoe de interne spanning reageert op de opgelegde rek. Het integreren van deze spanningsrespons levert het verschil in vrije energie tussen de twee fasen op.
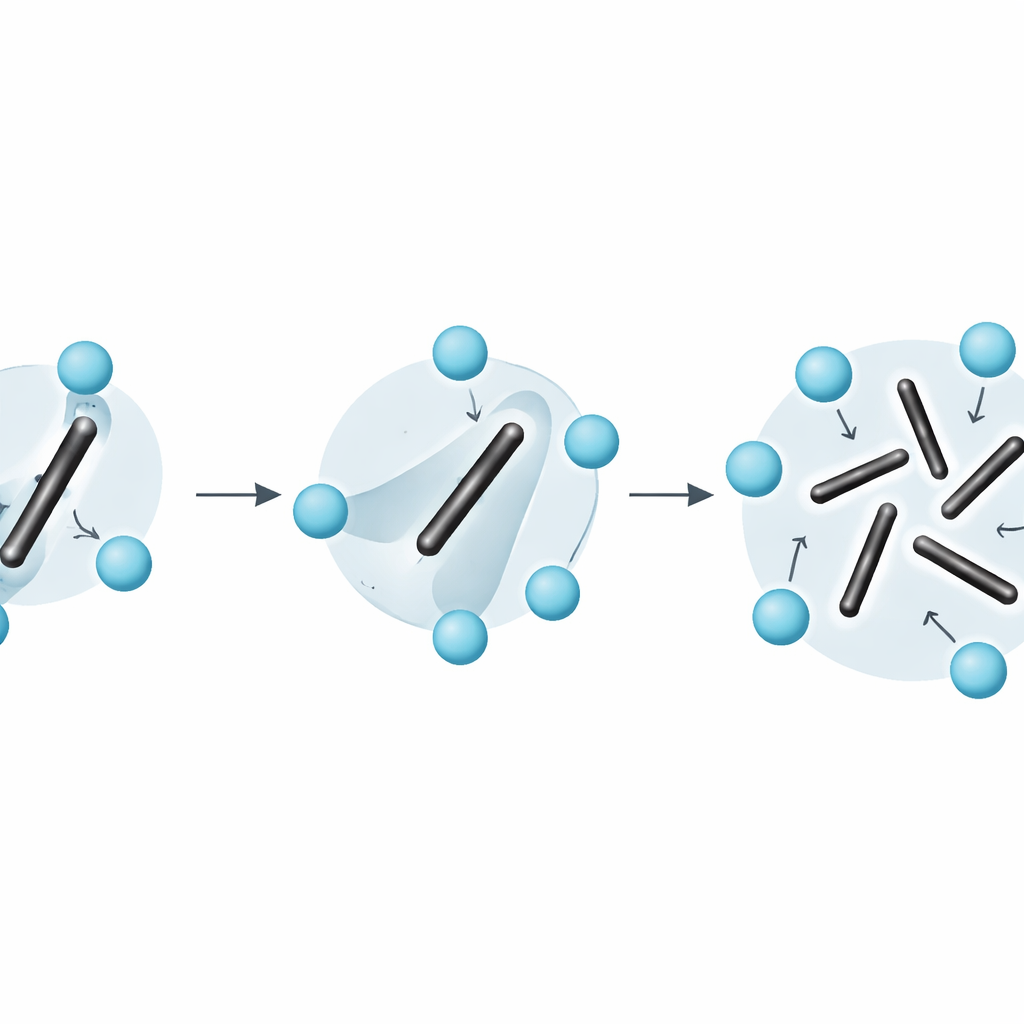
Entropie zien via atomaire beweging
De berekeningen laten zien dat rond 600 K de orthorombische fase bij lage temperatuur nog licht wordt bevoordeeld, terwijl bij 650 K de kubische fase wint met een paar duizendsten van een elektronvolt per formule-eenheid. Interpolatie tussen deze resultaten geeft een overgaanstemperatuur van ongeveer 611 K. Dit is lager dan experimentele schattingen maar nog steeds redelijk in overeenstemming, gezien de kleine vrije-energiedifferenties. De interne energie van de kubische fase is in feite hoger; wat deze fase stabiliseert is een grote entropiewinst, die rechtstreeks wordt herleid tot de rotatie-wanorde van de C2-dimeren. Door te analyseren hoe de oriëntatie van elke dimer in de tijd het geheugen van de begintoestand verliest, tonen de auteurs aan dat de dimeren zich heroriënteren op sub-picosecondeschaal en daarmee de scheidslijn vervagen tussen de gebruikelijke categorieën van "vibrationale" en "configuratie"-entropie.
Voorbij eenvoudige beelden van wanorde in vaste stoffen
Het werk benadrukt ook dat gangbare vereenvoudigingen — zoals het behandelen van entropie als een eenvoudige optelsom van vibraties rond vaste configuraties plus een afzonderlijke telling van statische oriënteringen — afbreken voor materialen zoals Li2C2. Omdat dimerrotaties snel zijn en sterk gekoppeld aan gewone vibraties, kan het systeem niet schoon worden opgesplitst in aparte "vibrerende" en "herordende" onderdelen. De stress–strain integratiemethode omzeilt deze moeilijkheid: ze haalt de volledige vrije energie direct uit de microscopische dynamica, zonder te hoeven raden hoe entropie verdeeld moet worden.
Wat de studie ons leert
In alledaagse bewoordingen laat de studie zien hoe een vaste stof rigide kan blijven terwijl zijn interne bouwstenen steeds vrijer worden om te draaien en te keren, en hoe deze interne vrijheid een meer gedesoriënteerde structuur thermodynamisch preferabel kan maken. Voor Li2C2 wordt de kubische fase bij hoge temperatuur niet gestabiliseerd omdat ze energetisch voordeliger is, maar omdat ze veel meer manieren biedt voor de C2-dimeren om zich te oriënteren en te bewegen. Door te bewijzen dat stress–strain thermodynamische integratie dit subtiele evenwicht tussen orde, energie en entropie kan vastleggen, baant het werk de weg naar het voorspellen van vergelijkbare overgangen in andere dynamisch gedesoriënteerde vaste stoffen die ten grondslag kunnen liggen aan toekomstige koelapparaten, batterijen en slimme materialen.
Bronvermelding: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Trefwoorden: vastestoffaseovergang, dynamische wanorde, moleculaire dynamica, lithiumcarbiden, thermodynamische integratie