Clear Sky Science · nl
Hybride bijgestemd deep learning-model voor borstkankerdiagnose op basis van genetische data
Waarom dit belangrijk is voor patiënten en families
Borstkanker is nu wereldwijd de meest gediagnosticeerde kanker bij vrouwen, en vroege opsporing kan het verschil betekenen tussen leven en dood. Artsen hebben steeds vaker toegang tot iemands genetische informatie, maar het omzetten van tienduizenden genmetingen in duidelijke antwoorden is buitengewoon moeilijk. Dit artikel beschrijft een nieuw computermodel dat deze complexe genetische patronen leest om borstkanker te detecteren en uitkomsten met opvallende nauwkeurigheid te voorspellen, en dat clinici mogelijk een krachtig hulpmiddel biedt voor eerdere en betrouwbaardere beslissingen.
Van genen naar waarschuwingssignalen
Elke borstkankertumor draagt een moleculair vingerafdruk die is gecodeerd in de activiteit van duizenden genen. De auteurs wilden een systeem bouwen dat deze vingerafdruk direct kan lezen, in plaats van alleen te vertrouwen op beeldvorming of een handvol bekende genen zoals BRCA1 en BRCA2. Ze werkten met twee van de grootste openbare bronnen in kanker-genomics: de TCGA-borstkankecohorte, die genactiviteit voor 17.814 genen in 590 monsters bevat, en de METABRIC-studie, die genomische en klinische informatie voor meer dan 1.400 patiënten omvat. Hun doel was ambitieus: een methode ontwerpen die deze informatievloed aankan, de meest betekenisvolle signalen vindt en toch betrouwbaar blijft in volledig onafhankelijke patiëntgroepen.
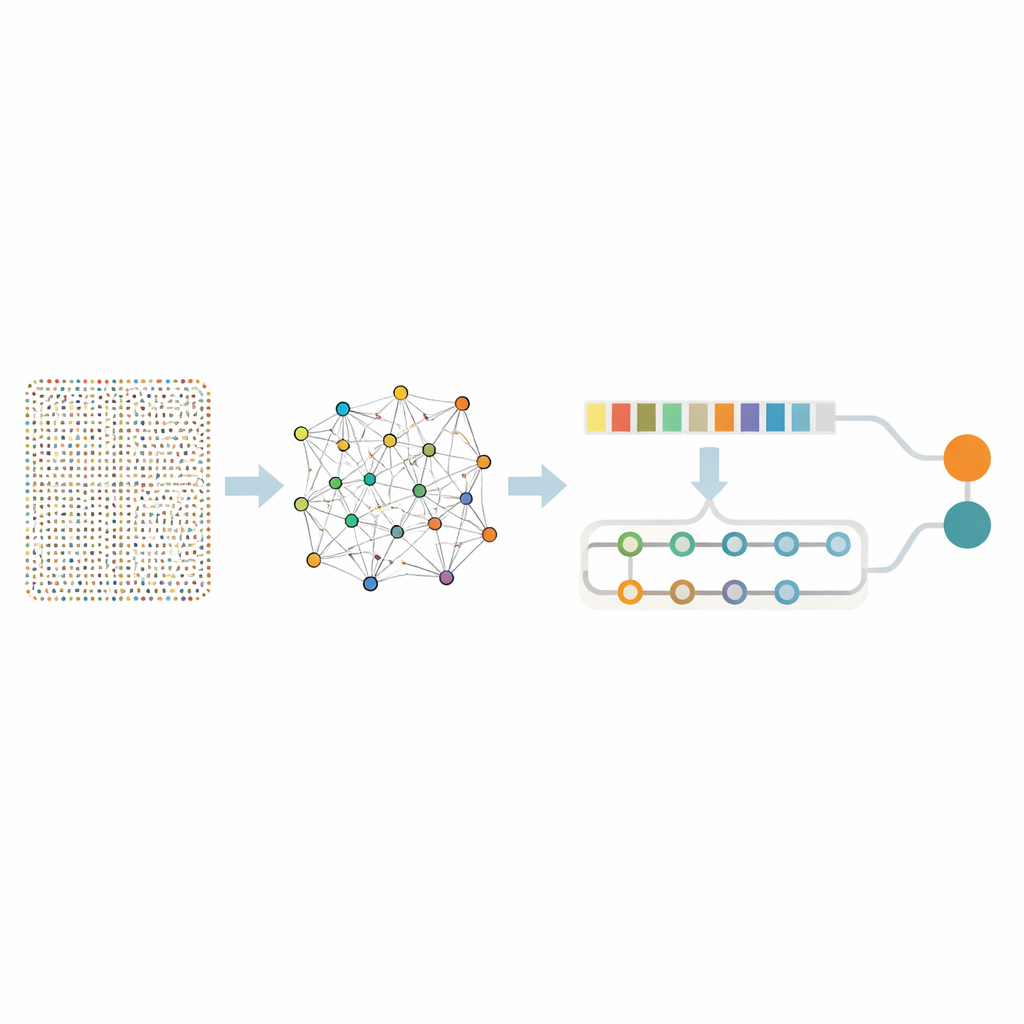
Duizenden genen terugbrengen naar een bruikbare set
Het tegelijk bekijken van bijna achttienduizend genen is overweldigend, zelfs voor geavanceerde algoritmen, en loopt het risico zinloze ruis te oppikken. De onderzoekers gebruikten daarom een tweeledige "zeef" om een kleinere set echt informatieve genen te isoleren. Eerst pasten ze een techniek toe die Random Forest heet, die effectief veel beslisbomen vraagt welke genen het belangrijkst zijn om kankerstekken van gezonde monsters te onderscheiden. Deze stap snoeide de lijst terug tot 436 veelbelovende genen. Vervolgens onderzochten ze hoe deze genen samen functioneren met behulp van association rule mining, een methode die genengroepen opspoort die de neiging hebben gelijktijdig actief te zijn in tumoren. Deze extra analysetrap identificeerde genparen en netwerken die verbonden zijn met sleutelprocessen van kanker zoals snelle celdeling, herstel van DNA-schade en veranderingen in het weefsel rond de tumor. Na deze verfijning bleven 332 genen over—nog steeds biologisch betekenisvol maar veel beter beheersbaar voor diepere analyse.
Een tweedelig neuraal netwerk dat patronen en context leert
Met deze gefocuste genenset bouwde het team een hybride deep learning-model dat twee typen neurale netwerken combineert. Het ene deel, bekend als een convolutioneel netwerk, scant langs de genlijst om lokale patronen op te pikken—clusters van genen die de neiging hebben gelijktijdig te stijgen of te dalen. Het tweede deel, een bidirectioneel geheugen-netwerk, bekijkt dezelfde informatie terwijl het langeafstandsrelaties bijhoudt en vastlegt hoe verre genen elkaar beïnvloeden over het gehele profiel. Voor het trainen balanceerden de auteurs de data zodat kanker- en niet-kanker-monsters eerlijk waren vertegenwoordigd en voegden ze kleine hoeveelheden kunstmatige ruis toe, om het model te leren zich niet te laten misleiden door toevallige fluctuaties.
Hoe goed het systeem presteert in real-world tests
Getraind en getest op TCGA-data onderscheidde het hybride netwerk tumor- van normale monsters correct met ongeveer 97% nauwkeurigheid en een bijna perfecte scheiding van de twee groepen. Belangrijk is dat het beter presteerde dan eenvoudigere deep learning-opstellingen en standaard machine learning-tools zoals logistieke regressie en support vector machines, zelfs wanneer die concurrerende methoden dezelfde zorgvuldig geselecteerde genen kregen. De sterkste toets was echter of het model standhoudt op een volledig andere dataset. Toegepast op METABRIC, dat in andere ziekenhuizen met andere laboratoriummethoden werd verzameld, behield het systeem hoge prestaties: in zijn beste run behaalde het 99,3% nauwkeurigheid en identificeerde het correct elke patiënt die later aan borstkanker stierf, een cruciale eigenschap als het hulpmiddel gebruikt zou worden om hoog-risico gevallen te signaleren.
Wat dit zou kunnen betekenen voor toekomstige zorg
Voor niet-specialisten komt het erop neer dat deze studie een slimme filter en lezer voor genetische data biedt die borstkanker en aanverwant risico met opmerkelijke consistentie kan opsporen in grote patiëntengroepen. Door een doordachte genselectiestrategie te combineren met een tweetakkig neuraal netwerk laten de auteurs zien dat computers klinisch betekenisvolle signalen uit enorme genetische datasets kunnen halen, niet alleen in één studie maar over onafhankelijke cohorten heen. Hoewel meer werk nodig is om de aanpak in diverse populaties te testen en de beslissingen ervan gedetailleerd te verklaren, wijst de methode op een toekomst waarin een eenvoudig bloed- of weefselmonster zulke modellen kan voeden en artsen kan helpen tumoren eerder te detecteren en behandelingen nauwkeuriger af te stemmen.
Bronvermelding: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Trefwoorden: borstkanker-genomics, deep learning-diagnose, genexpressie-biomarkers, vroege kankerdetectie, klinische besluitvorming ondersteuning