Clear Sky Science · nl
DPAS: disease-associated peptide anomaly score voor het identificeren van pathogene peptiden via one-class learning
Waarom kleine eiwitfragmenten belangrijk zijn voor onze gezondheid
Peptiden—korte stukken van eiwitten—hebben zich ontpopt tot belangrijke spelers in de moderne geneeskunde. Ze kunnen als precieze boodschappers in het lichaam fungeren en worden steeds vaker gebruikt als geneesmiddelen en ziektemarkers. Toch vereist het bepalen welke peptiden daadwerkelijk aan ziekte gekoppeld zijn doorgaans duidelijke voorbeelden van zowel “ziekte” als “niet‑ziekte” peptiden, iets wat de biologie zelden biedt. Deze studie introduceert een nieuwe manier om potentieel schadelijke peptiden te ontdekken met alleen de peptiden waarvan we al weten dat ze bij ziekte betrokken zijn, wat een snellere en minder bevooroordeelde route biedt naar toekomstige diagnostiek en behandelingen.
De uitdaging van het vinden van de “niet‑ziekte” groep
Traditionele computermodellen leren door twee kanten te vergelijken: positieve voorbeelden die bekend zijn als ziektegerelateerd en negatieve voorbeelden die als onschadelijk worden beschouwd. In peptidenonderzoek vormt die tweede groep een probleem. Veel peptiden zijn simpelweg niet getest, dus ze bestempelen als “niet‑ziekte” kan misleidend zijn en bias introduceren. Vorige studies naar anti‑kanker of anti‑inflammatoire peptiden bereikten indrukwekkende nauwkeurigheid, maar vertrouwden vaak op handmatig samengestelde of veronderstelde negatieve datasets. Daardoor kunnen hun modellen moeite hebben met zeldzame signalen of nieuwe typen ziektepeptiden die niet op het trainingsmateriaal lijken.
Leren van wat we weten in plaats van wat we veronderstellen
De auteurs kiezen een andere aanpak: in plaats van er een tweezijdig probleem van te maken, behandelen ze ziektegeassocieerde peptiden als één samenhangende groep en vragen ze: “Hoe ziet deze groep er precies uit?” Ze verzamelen meer dan 760.000 gemuteerde menselijke peptiden uit een gespecialiseerde kankergerelateerde database en beschrijven elk peptide met een rijke set kenmerken. Daarbij gaat het onder meer om hoe vaak elk aminozuur voorkomt, hoe paren van aminozuren gerangschikt zijn, basale fysisch‑chemische eigenschappen zoals volume en hydrofiele/neutrale eigenschappen, en korte terugkerende sequentiepatronen die motieven worden genoemd. Een techniek genaamd principal component analysis comprimeert deze veel-dimensionale beschrijving tot een beter hanteerbare vorm, terwijl de belangrijkste variatiebronnen behouden blijven.
Ongewone peptiden opsporen met one-class modellen
Met deze gecomprimeerde kenmerkenruimte trainen de onderzoekers drie “one‑class” modellen—algoritmen die zijn ontworpen om de vorm van één groep te leren en afwijkende voorbeelden te markeren. Ze testen One‑Class Support Vector Machines, Isolation Forests en een type neuraal netwerk dat een autoencoder wordt genoemd. De autoencoder leert om elk peptidekenmerk samen te persen naar een smalle interne representatie en het daarna te reconstrueren; peptiden die tot het aangeleerde ziektepatroon behoren worden nauwkeurig herbouwd, terwijl ongebruikelijke peptiden een hogere reconstructiefout opleveren. Vergelijking van genormaliseerde anomaliescores over alle methoden laat zien dat de autoencoder de meest compacte cluster van typische peptiden produceert en de duidelijkste scheiding tussen inliers en outliers. Door een drempel te zetten op reconstructiefout rond het 95e percentiel classificeert het model de meerderheid van de peptiden als waarschijnlijk ziektegeassocieerd, terwijl consequent een klein deel als atypisch wordt gemarkeerd.
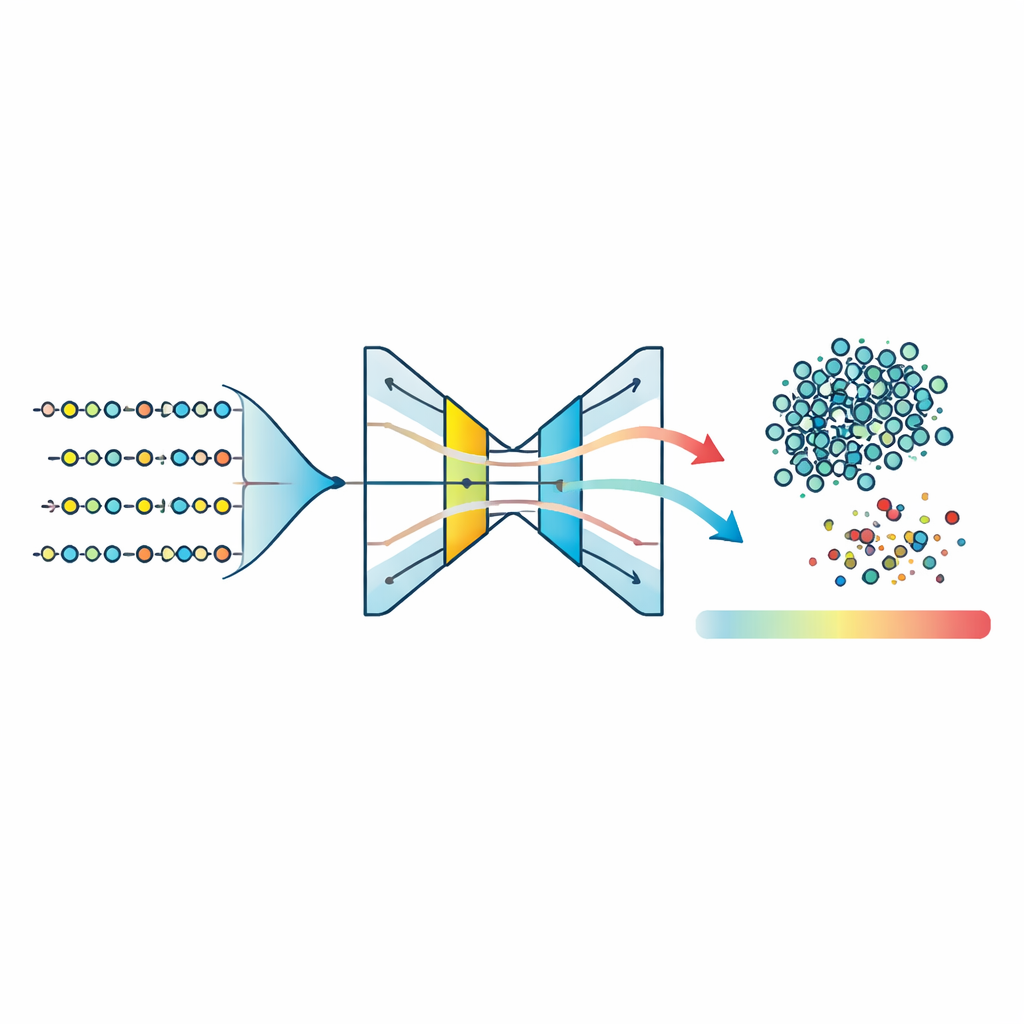
Complexe scores terugbrengen tot één zinvol getal
Om de resultaten biologisch beter interpreteerbaar te maken, introduceren de auteurs de Disease Peptide Anomaly Score (DPAS). Deze score mengt twee bestanddelen: hoe ongewoon een peptide eruitziet voor de autoencoder (de genormaliseerde reconstructiefout) en hoe sterk zijn kenmerken bijdragen aan voorspellingen, gemeten met een populaire verklaringsmethode genaamd SHAP. In de praktijk blijken motieven en specifieke fysisch‑chemische eigenschappen bijzonder informatief te zijn. DPAS combineert deze signalen zodat peptiden die zowel structureel afwijkend zijn als ondersteund worden door biologisch betekenisvolle kenmerken hogere rang krijgen. De hoogst scorende peptiden worden vervolgens onderzocht met een motiefzoektool die ze koppelt aan bekende functionele signaturen zoals fosforyleringsplaatsen, metaalbindende regio’s en andere regulatoire patronen die vaak betrokken zijn bij signalering en enzymcontrole.
Wat dit betekent voor toekomstige diagnostiek en geneesmiddelen
Concreet biedt dit werk een slimmer filter om verdachte peptiden te vinden zonder te doen alsof we weten welke peptiden zeker onschadelijk zijn. Door alleen te leren van bevestigde ziektegerelateerde voorbeelden en nieuwe kandidaten met DPAS te rangschikken, kunnen onderzoekers een korte, biologisch plausibele lijst peptiden prioriteren voor laboratoriumtests. Veel van de hoogst gerangschikte kandidaten bevatten bekende functionele motieven, wat de suggestie versterkt dat ze mogelijk een rol spelen in ziekteprocessen. Hoewel de methode nog afhankelijk is van aannames en experimenteel bewezen “veilige” peptiden voor volledige validatie mist, biedt zij een realistischer en transparanter fundament voor de ontdekking van peptiden‑biomarkers en kan ze worden aangepast aan andere biologische datatypen waar betrouwbare negatieve voorbeelden schaars zijn.
Bronvermelding: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Trefwoorden: peptiden geassocieerd met ziekte, anomaliedetectie, autoencoder, biomarkerontdekking, one-class learning