Clear Sky Science · nl
Interpreteerbare machine learning verklaart remming van carbonische anhydrase via conformele en tegenfeitelijke voorspellingen
Waarom slimmer gemaakte kankergeneesmiddelen ertoe doen
Kankergeneesmiddelen werken vaak als botte instrumenten: ze vallen tumorcellen aan, maar kunnen ook gezonde weefsels beschadigen en ernstige bijwerkingen veroorzaken. Een veelbelovende manier om die precisie te verbeteren is het blokkeren van specifieke vormen van een enzym genaamd carbonische anhydrase, dat tumoren helpt overleven in zuurstofarme omgevingen. Verschillende vormen van dit enzym lijken echter bijna identiek, waardoor het moeilijk is geneesmiddelen te ontwerpen die de “slechte” tumorenversies treffen zonder de “goede” vorm in het lichaam te verstoren. Deze studie laat zien hoe interpreteerbare machine learning onderzoekers kan helpen deze uitdaging te navigeren en selectievere, veiligere medicijnkandidaten te ontwerpen.
Het probleem van het raken van het verkeerde doelwit
Humane carbonische anhydrase (hCA) komt in vele vormen, of isoformen. Twee daarvan, IX en XII, zijn gekoppeld aan het overleven van kankercellen in zuurstofarme tumoren, dus het blokkeren ervan kan de ziekte vertragen en de behandeling verbeteren. Maar isoform II is wijdverspreid in gezonde weefsels en heeft een actief centrum dat sterk lijkt op dat van IX en XII. Geneesmiddelen die aan alle drie binden kunnen ongewenste problemen uitlokken, zoals metabole acidose en gezichtsstoornissen. Traditionele laboratorium- en rekenmethoden hebben moeite omdat enzymen grote, complexe moleculen zijn en het aantal mogelijke geneesmiddelachtige verbindingen astronomisch is. Alles uitputtend testen, zowel in het lab als in silico, is simpelweg niet haalbaar.
Het bouwen van een schone en betrouwbare gegevensbasis
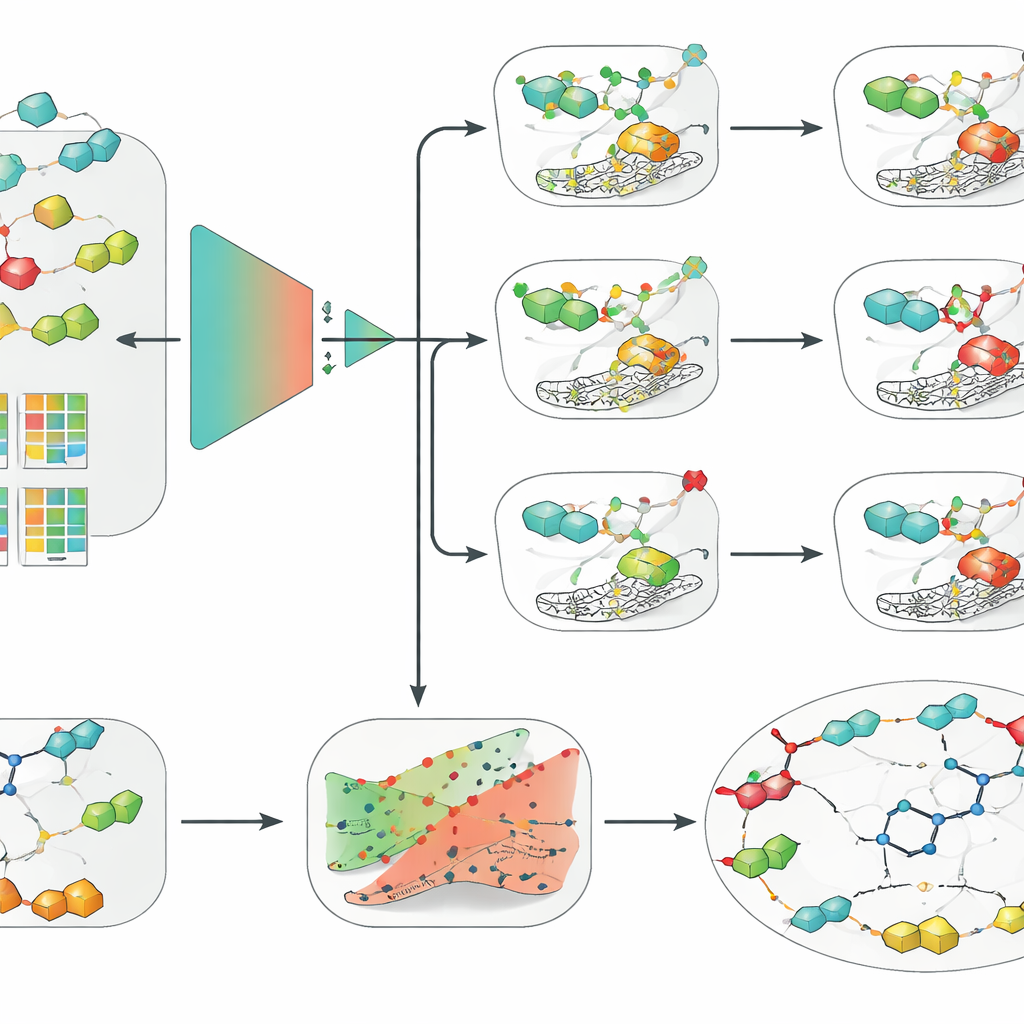
De auteurs pakten dit aan door eerst een zorgvuldig schoongemaakte databank samen te stellen van duizenden moleculen getest tegen hCA II, IX en XII vanuit de ChEMBL-repository. Ze standaardiseerden chemische structuren, verwijderden dubieuze metingen en richtten zich op verbindingen die een gemeenschappelijke zinkbindende groep gedeeld door deze klasse remmers bevatten. Met strikte drempels labelden ze moleculen als duidelijk actief of duidelijk inactief en verwijderden grijsgevallen die de modellen konden verwarren. Omdat er veel meer inactieve dan actieve moleculen waren, balanceerden ze de data zodat leeralgoritmen niet eenvoudigweg de meerderheidsgroep zouden bevoordelen. Ze gebruikten ook een ‘scaffold-gebaseerde’ manier om de data te splitsen, zodat trainings- en testsets verschillende kernmoleculaire raamwerken bevatten — wat een realistischer beeld geeft van hoe goed de modellen echt nieuwe verbindingen aankunnen.
Eenvoudige modellen verslaan deep learning wanneer data beperkt zijn
Met deze gecureerde dataset vergeleek het team een breed scala aan benaderingen, van klassieke machine learning-methoden zoals logistieke regressie, random forests en support vector machines (SVM) tot moderne diepe neurale netwerken, inclusief grafgebaseerde modellen die direct op moleculaire structuren werken. Ze combineerden deze met verschillende manieren om moleculen te coderen, zoals traditionele handgemaakte descriptors, sleutel-gebaseerde fingerprints en geleerde embeddings uit een chemisch taalmodel. Over alle drie de enzymisoformen en onder de strengere scaffold-gebaseerde evaluatie stak één combinatie consequent bovenuit: een SVM gevoed met extended-connectivity fingerprints, een gestructureerde manier om lokale chemische omgevingen binnen een molecuul te beschrijven. Vreemd genoeg overtrof deze relatief eenvoudige opzet trendier graf- en deep learning-modellen, wat benadrukt dat datakwaliteit, zorgvuldige validatie en goede moleculaire descriptors meer kunnen wegen dan algoritmische complexiteit wanneer datasets van bescheiden omvang zijn.
Betrouwbaar vertrouwen en mensvriendelijke verklaringen toevoegen
De onderzoekers wikkelden hun beste SVM-model vervolgens in twee extra lagen die bedoeld waren om de voorspellingen bruikbaarder te maken voor echte geneesmiddelontdekking. Ten eerste pasten ze een raamwerk toe dat conformele voorspelling heet, dat niet slechts een ja-of-nee-antwoord geeft maar in plaats daarvan een bereik van waarschijnlijke uitkomsten met een gegarandeerde foutkans levert. Dit stelt wetenschappers in staat te bepalen hoe voorzichtig ze willen zijn en gevallen te herkennen waarin het model echt onzeker is. Ten tweede gebruikten ze tegenfeitelijke verklaringen om de redenering van het model intuïtiever te maken. Voor een gegeven molecuul genereerden ze nauw verwante analogen die de voorspelde uitkomst doen omslaan van actief naar inactief, of andersom. Het onderzoeken van deze paren voor de klinische kandidaat SLC-0111, die selectief IX en XII blokkeert maar niet II, herontdekte de methode onafhankelijk een belangrijk inzicht uit de medicinale chemie: kleine veranderingen in het “staart”-gedeelte van het molecuul veranderen sterk welke isoform het bij voorkeur bindt.
Van algoritmes naar praktische hulpmiddelen voor geneesmiddelenontwerp
Om hun benadering toegankelijk te maken, verpakten de auteurs de drie SVM-modellen, de onzekerheidslaag en de tegenfeitelijke motor in een grafische tool genaamd CAInsight. Een gebruiker kan de tekstuele representatie van een molecuul opgeven en met één klik voorspelde activiteit tegen hCA II, IX en XII ophalen, een schatting van hoe betrouwbaar elke voorspelling is, en voorgestelde structurele aanpassingen die activiteit kunnen vergroten of verminderen. Hoewel de modellen zich richten op het classificeren van moleculen als actief of inactief in plaats van het in één stap voorspellen van exacte potentie of selectiviteit, reproduceren ze al bekend gedrag voor echte geneesmiddelkandidaten en onderscheiden ze subtiele structurele veranderingen. De auteurs merken op dat grotere en meer uniforme datasets, plus een diepere analyse van hoe activiteitsdrempels gekozen worden, de prestaties verder kunnen verfijnen.
Wat dit betekent voor toekomstige kankergeneesmiddelen
In eenvoudige bewoordingen laat dit werk zien dat zorgvuldig opgebouwde en goed verklaarde machine learning-modellen chemici kunnen helpen kankergeneesmiddelen te ontwerpen die beter onderscheid maken tussen sterk gelijkende enzymdoelen. Door robuuste statistiek, onzekerheidsschattingen en intuïtieve “wat-als”-voorbeelden te combineren, voorspelt het kader niet alleen welke moleculen waarschijnlijk werken, maar suggereert het ook waarom. Dit soort transparante kunstmatige intelligentie kan virtuele screening versnellen, generatief ontwerp van nieuwe verbindingen ondersteunen en de trial-and-error last in het lab verminderen, wat uiteindelijk kan bijdragen aan de ontdekking van selectievere en veiligere behandelingen voor patiënten.
Bronvermelding: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Trefwoorden: remmers van carbonische anhydrase, interpreteerbare machine learning, drugselectiviteit, conforme voorspelling, tegenfeitelijke verklaringen