Clear Sky Science · nl
Compound heterozygote CHAT-genmutaties, een missense- en een splice-sitevariant, bij twee broers en zussen met congenitaal myastheniesyndroom
Wanneer ademhalen plotseling faalt
Sommige kinderen lijken bij de geboorte gezond maar stoppen tijdens geringe koortsen plotseling met ademhalen en hebben dan noodopvang met beademing nodig. Voor hun families zijn die aanvallen angstaanjagend en raadselachtig. Deze studie onderzoekt twee dergelijke broers en zussen uit Japan en herleidt hun levensbedreigende aanvallen van zwakte en apneu (ademhalingsstilstanden) tot kleine veranderingen in één gen dat zenuwen helpt met spieren te communiceren. Door klinische aanwijzingen, genoomsequencing en computergebaseerde eiwitmodellering te combineren tonen de onderzoekers hoe deze mutaties waarschijnlijk een belangrijk enzym verstoren en artsen een duidelijker doel geven voor diagnose en behandeling.
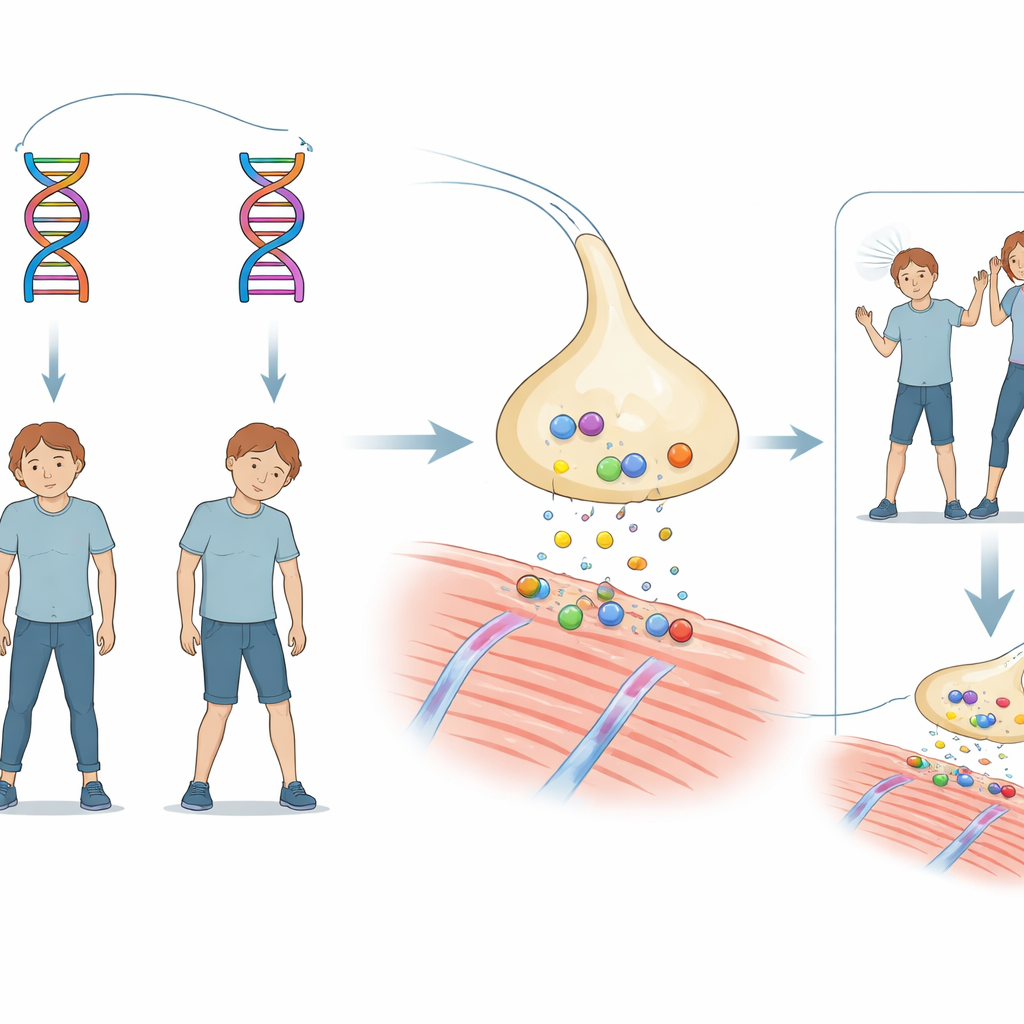
Een familieberoerte van plotselinge zwakte
Het verhaal draait om een broer en zus die beiden in hun vroege kinderjaren iets vertraagde motorische ontwikkeling hadden. Rond 18 maanden kregen beide kinderen aanvallen van apneu en bewustzijnsverlies tijdens koorts, ernstig genoeg om beademing te vereisen. Toen ze ouder werden, hadden ze beiden terugkerende loodrechte oogleden (ptosis) en algemene spierzwakte die werd uitgelokt door infecties, koorts of inspanning. Hersenonderzoeken waren normaal en de veelvoorkomende antilichaamgedreven vormen van myasthenie (een ziekte waarbij de communicatie tussen zenuw en spier is verstoord) werden uitgesloten. Toch verbeterden hun klachten duidelijk met een geneesmiddel dat het chemische signaal tussen zenuw en spier versterkt, wat wees op een zeldzame erfelijke aandoening genaamd congenitaal myastheniesyndroom.
Het vinden van de foutieve instructies
Om een erfelijke oorzaak te zoeken, sequentieerden de onderzoekers alle eiwitcoderende genen van de twee kinderen en hun ouders. Ze ontdekten dat elk kind twee verschillende veranderingen in hetzelfde gen, CHAT, droeg; dit gen codeert voor choline-acetyltransferase — een enzym dat acetylcholine maakt, de belangrijkste chemische boodschapper waarmee zenuwen spieren activeren. De ene wijziging veranderde een enkele bouwsteen van het enzym (een missense-mutatie bekend als G411R). De andere bevond zich op een cruciale grens waar de cel normaal gesproken gensegmenten knipt en plakt tijdens de RNA-vorming (een splice-sitemutatie aangeduid als c.752+2T>C). Elke ouder droeg slechts één van deze veranderingen en was gezond; alleen de kinderen die beide veranderingen erfden, vertoonden ziekte, wat suggereert dat de combinatie van mutaties samen de enzymfunctie verzwakt.
Onderzoeken hoe een verborgen knip het enzym verandert
Aangezien de onderzoekers niet genoeg natuurlijk CHAT-RNA uit bloedcellen konden verkrijgen, gebruikten ze een "minigene"-experiment. Ze klaarden het relevante gedeelte van het gen in een DNA-vector, introduceerden ofwel de normale of de gemuteerde versie in gekweekte cellen en onderzochten vervolgens hoe het RNA werd verwerkt. In de normale constructie bevatte het RNA alle verwachte segmenten. In de gemuteerde versie werd een heel segment dat bekendstaat als exon 5 overgeslagen, maar het leesraam van het gen bleef intact. Dit betekende dat het enzym zou worden aangemaakt maar een korte interne reeks aminozuren zou missen. Evolutionaire vergelijkingen toonden aan dat dit ontbrekende gebied sterk geconserveerd is tussen soorten, wat erop wijst dat het een belangrijke structurele rol vervult.
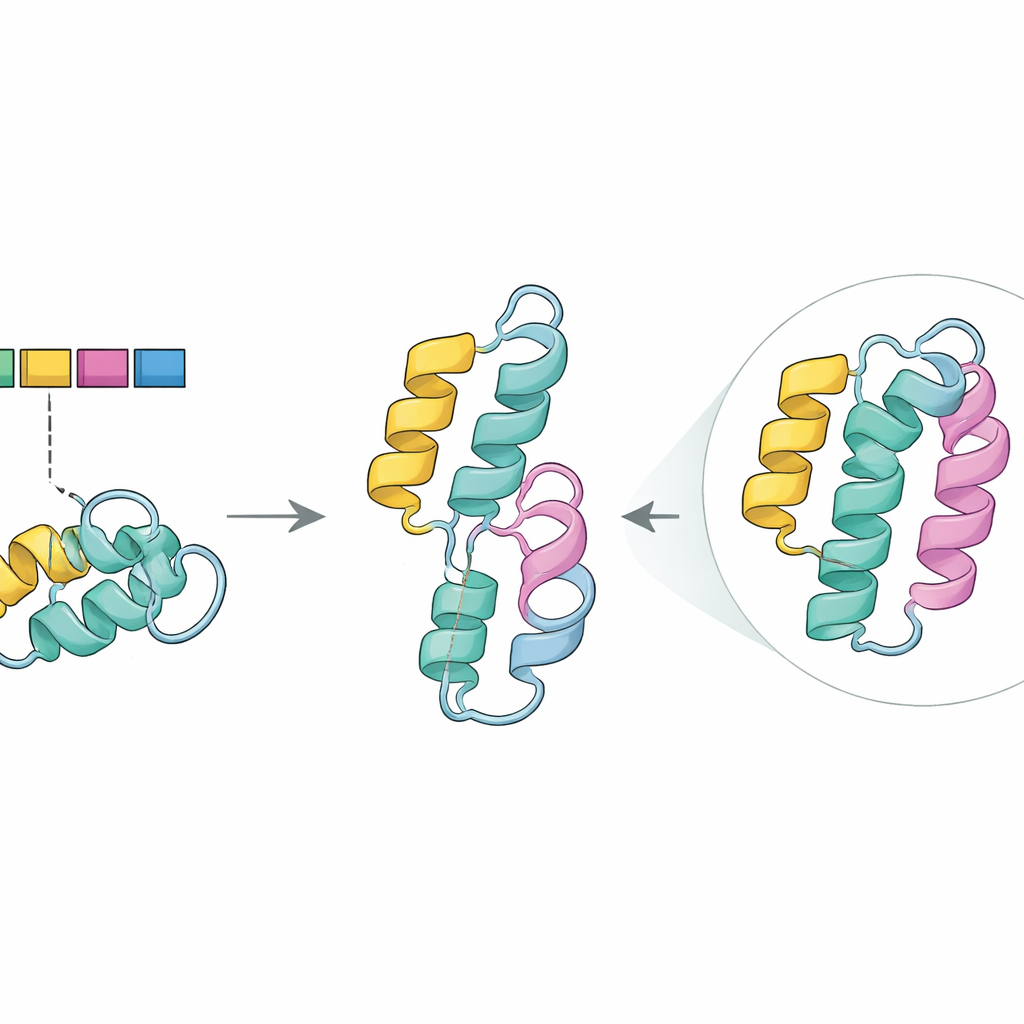
Structurele schade in silico waarnemen
Om die rol te onderzoeken, schakelde het team AlphaFold2 in, een geavanceerd programma dat driedimensionale eiwitstructuren voorspelt op basis van sequenties. In het normale enzym vormt het door exon 5 gecodeerde deel een van de strak opgerolde spiraalsegmenten (een alfahelix) die helpen het kerngebied van het eiwit te stabiliseren. In de voorspelde gemuteerde structuur verdween deze helix, waardoor een kloof ontstond in een regio die uit eerder werk bekend is als cruciaal voor het behouden van stabiliteit en het ondersteunen van effectieve katalyse. Samen met computertools die schadelijke mutaties aanwijzen, ondersteunen deze resultaten het idee dat het overslaan van exon 5, vooral in combinatie met de G411R-verandering op het andere genkopie, de prestaties van het enzym ondermijnt zonder het volledig uit te schakelen — in overeenstemming met de matige maar ernstige klachten van de broers en zussen.
Wat dit betekent voor patiënten en families
De studie concludeert dat de combinatie van de G411R-missensemutatie en de nieuw geïdentificeerde splice-sitemutatie in CHAT zeer waarschijnlijk verantwoordelijk is voor het congenitaal myastheniesyndroom bij de twee kinderen. Door met de minigene-assay en structurele modellering aan te tonen hoe de splice-siteverandering een stabiliserende helix uit het enzym verwijdert, leveren de auteurs een mechanistische verklaring waarop clinici en onderzoekers kunnen voortbouwen. Voor getroffen families betekent dit werk meer dan alleen een naam: het ondersteunt gerichte behandeling met geneesmiddelen die de neuromusculaire signalering versterken, begeleidt genetische counseling bij toekomstige zwangerschappen en voegt een belangrijk nieuw voorbeeld toe aan het register van hoe subtiele veranderingen in onze genetische code de spierkracht en de fundamentele handeling van ademhalen diepgaand kunnen beïnvloeden.
Bronvermelding: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Trefwoorden: congenitaal myastheniesyndroom, CHAT-gen, choline-acetyltransferase, splice-sitemutatie, neuromusculaire overgang