Clear Sky Science · nl
Aandachtsgestuurde verbeterde deconvolutie maakt referentievrije schatting van celtypen in ruimtelijke transcriptoomica mogelijk
Cellen in hun context zien
Moderne biologie kan de activiteit van duizenden genen tegelijk aflezen, niet alleen in geïsoleerde cellen maar direct binnen dunne weefselsneden. Dit "ruimtelijke transcriptoomica"-perspectief onthult waar verschillende cellen zich bevinden en hoe ze interageren, maar elke meting mengt vaak signalen van veel naburige cellen. De studie introduceert een nieuwe computationele methode, AGED genoemd, die deze mengsels kan ontwarren en schat welke celtypen waar aanwezig zijn—zonder een apart, zorgvuldig afgestemd single-cell referentie-dataset te hoeven gebruiken. 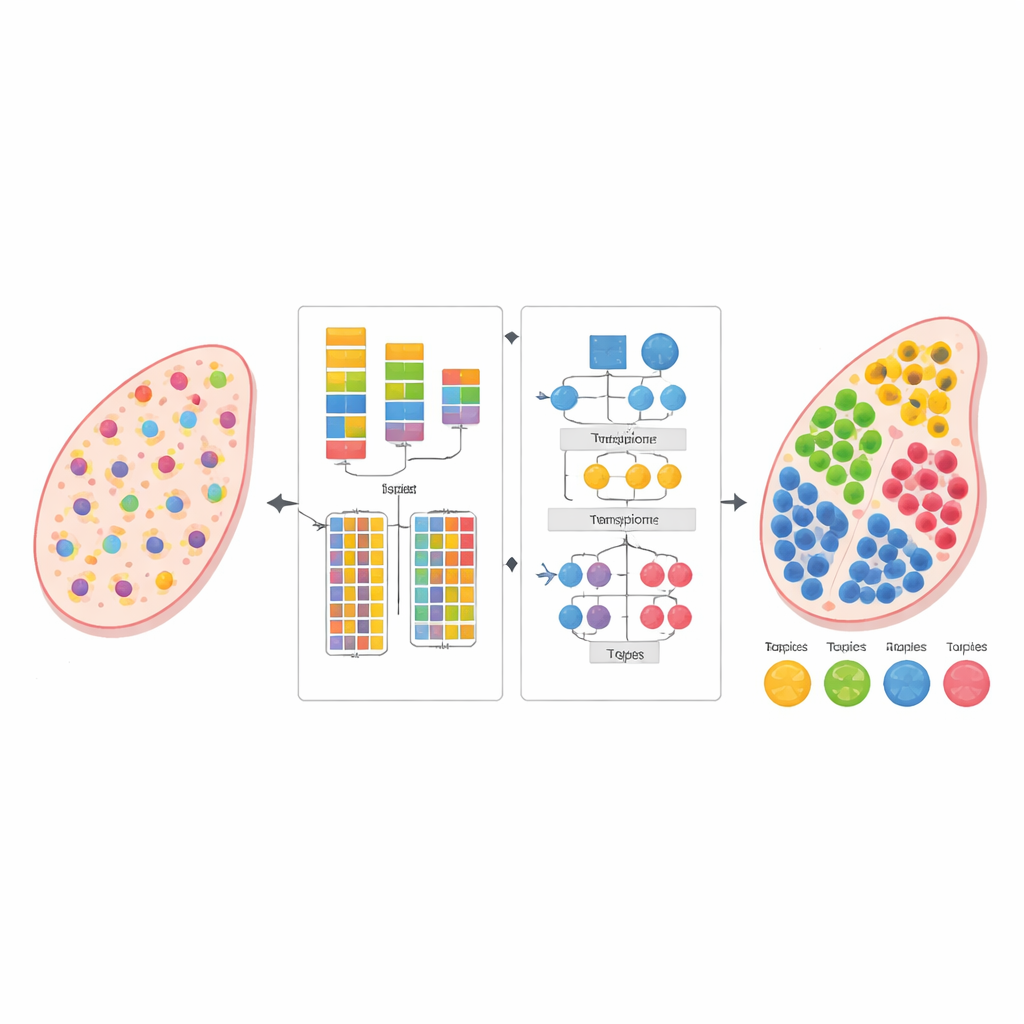
Waarom cellen in weefsels in kaart brengen moeilijk is
Platforms voor ruimtelijke transcriptoomica meten genactiviteit over een raster van plekjes die over een weefselsnede liggen. Omdat de meeste plekjes meerdere cellen tegelijk vastleggen, moeten onderzoekers de gemengde signalen wiskundig ontleden om de onderliggende celtypen en hun verhoudingen te herstellen. Bestaande tools vertrouwen vaak op externe single-cell referentieatlassen van hetzelfde weefsel. Die atlassen kunnen ontbreken voor zeldzame weefsels, speciale ziektebeelden of ongebruikelijke experimentele condities, en zelfs wanneer ze beschikbaar zijn, sluiten ze mogelijk niet perfect aan, wat vertekening kan introduceren. Referentievrije methoden vermijden deze afhankelijkheid, maar huidige benaderingen hebben moeite met complexe ruimtelijke patronen, subtiele genrelaties en de uitdaging om te bepalen hoeveel verschillende celtypen überhaupt gezocht moeten worden.
Een tweestapsstrategie om mengsels te ontwarren
De auteurs ontwierpen AGED als een tweefasig kader dat ideeën uit de statistiek en modern deep learning combineert. In de eerste fase test de methode een reeks mogelijkheden voor hoeveel celtypen in het weefsel aanwezig zouden kunnen zijn. Ze gebruikt een snelle attention-gebaseerde neurale netwerkarchitectuur, bekend als een Performer, om kandidaatdecomposities te leren en scoort die vervolgens met meerdere criteria tegelijk: hoe goed het model de waargenomen genaantallen reconstrueert, hoe duidelijk de afgeleide celgroepen van elkaar gescheiden zijn en hoe divers die groepen zijn. Een curve-fitprocedure vindt een "elbow point" waar het toevoegen van meer celtypen weinig voordeel oplevert, waardoor de methode automatisch een geschikt aantal kan selecteren in plaats van te vertrouwen op een gok van de gebruiker.
Aangegane aandacht om biologie vast te leggen
Zodra het aantal celtypen is vastgesteld, verfijnt AGED in de tweede fase de oplossing met een rijkere attention-gebaseerde architectuur. Het begint bij een statistisch topicsmodel dat elk weefselplekje behandelt als een mengsel van verborgen "thema’s"—hier staand voor celtypen—en elk celtype als een karakteristiek genpatroon. Deze initiële thema’s bieden globale structuur. Het model legt vervolgens meerdere aandachtmechanismen bovenop: één koppelt de statistische thema’s aan het neurale netwerk, een andere verzamelt informatie van naburige plekjes in de fysieke ruimte, en een derde verbindt thema’s direct met genen. Een gate-systeem laat het model per geval beslissen hoeveel vertrouwen te hechten aan de voorafgaande statistische patronen versus de lokale data. Extra beperkingen stimuleren spaarzame oplossingen, wat de biologische realiteit weerspiegelt dat de meeste weefsellocaties gedomineerd worden door slechts een paar hoofdceltypen. 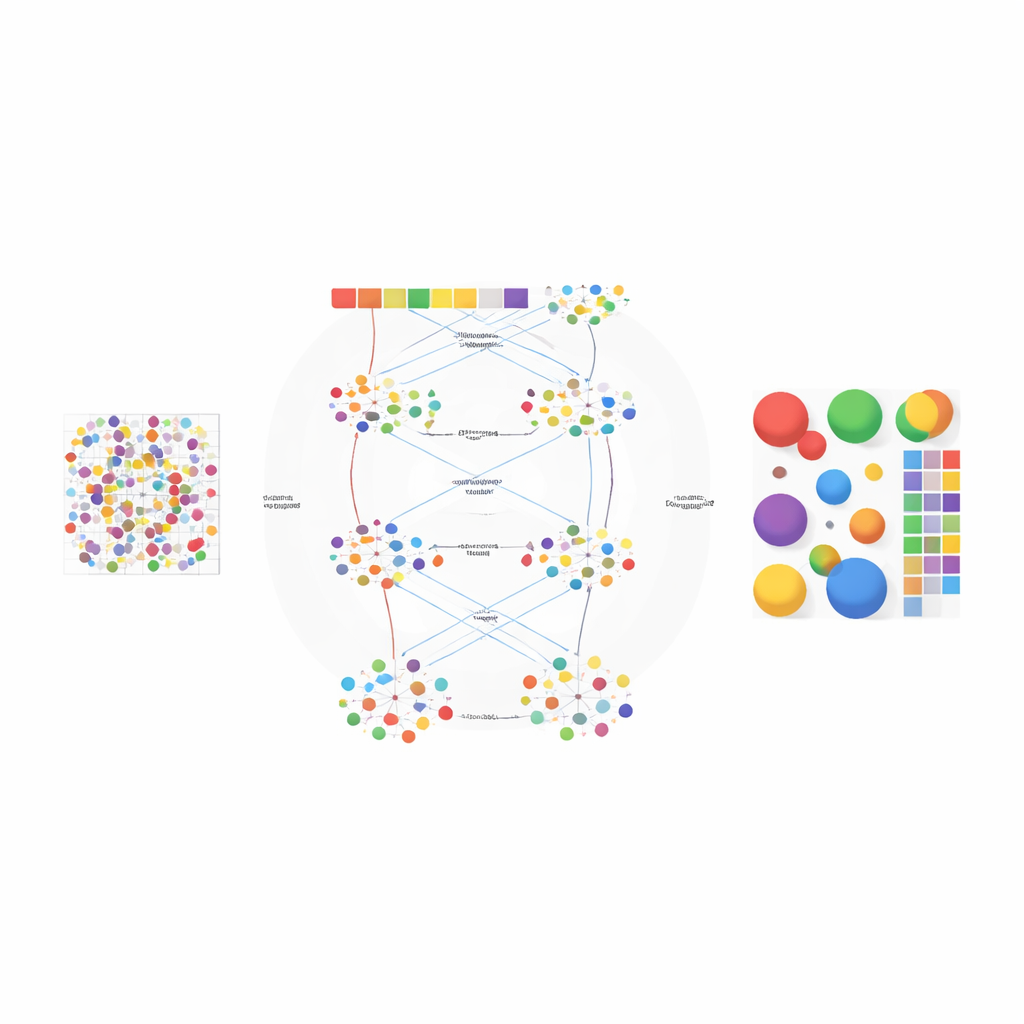
De methode op de proef stellen
De onderzoekers evalueerden AGED op meerdere soorten data. In gesimuleerd muizengeurkolbomweefsel herstelde de methode vier bekende anatomische lagen en kwam de geschatte celcomposities dichter bij de werkelijke samenstellingen dan veelgebruikte referentiegebaseerde en referentievrije tools, met zowel hoge correlatie met de grondwaarheid als lage reconstructiefout. In humaan pancreatisch ductaal adenocarcinoom koos AGED automatisch een oplossing met twintig celtypen die overeenkwam met door pathologen geannoteerde regio’s zoals tumor, afvoerbuis en normaal pancreasweefsel, en overtrof andere methoden op een structurele similariteitsmaat die de afgeleide kaarten vergelijkt met de zichtbare weefselstructuur. In humaan thymusweefsel scheidde AGED nauwkeurig belangrijke celpopulaties en legde het een biologisch verwachte negatieve relatie vast tussen twee gespecialiseerde epitheelceltypen—een patroon dat concurrerende benaderingen niet wisten te reproduceren. Aanvullende analyses op andere datasets en op resoluties die op single-cell niveau lijken, ondersteunden verder de robuustheid van de methode.
Wat dit vooruit betekent
Voor niet-specialisten kan AGED worden gezien als een slimme ontmengingmotor voor complexe weefsels: het leert hoeveel verschillende celgemeenschappen aanwezig zijn, waar ze zich bevinden en welke genen ze definiëren, alles alleen uit de ruimtelijke data. Door interpreteerbare statistische modellen te verweven met flexibele attention-gebaseerde neurale netwerken biedt het kader zowel nauwkeurigheid als inzicht, zelfs wanneer geen geschikte referentieatlas beschikbaar is. Dit maakt het een praktisch instrument om weefselorganisatie te verkennen in gezondheid en ziekte, van hersenlagen tot tumoren en immuunorganen, en wijst op een bredere strategie om voorafgaande kennis te gebruiken om krachtige maar ondoorzichtige machine-learningmodellen in de biologie te sturen.
Bronvermelding: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Trefwoorden: ruimtelijke transcriptoomica, deconvolutie van celtypen, deep learning, weefselarchitectuur, referentievrije analyse