Clear Sky Science · nl
LncRNA FTX bevordert myocardiale fibrose door miR-335-3p te sponsoren en zo het TFEC/ILK-signaal te reguleren
Waarom littekenvorming in het hart ertoe doet
Hartfalen treft tientallen miljoenen mensen wereldwijd en sluipt vaak jarenlang onopgemerkt voort. Een belangrijke oorzaak van deze achteruitgang is myocardiale fibrose—langzame, progressieve littekenvorming in de hartspier waardoor die stijver wordt en minder goed bloed kan pompen. Deze studie onderzoekt de moleculaire "bedrading" die hartcellen aanzet om te veel littekenweefsel aan te maken en identificeert een nieuwe keten van moleculen die mogelijk kan worden gericht om dit schadelijke proces te vertragen of zelfs om te keren.
Een nadere blik op hartlittekenvorming
Wanneer het hart beschadigd raakt of onder stress staat, komen ondersteunende cellen genaamd cardiale fibroblasten in actie. Bij gezonde reparatie helpen zij schade te herstellen. Bij chronische ziekte kunnen ze echter in een overactieve staat overschakelen en te veel collageen en andere componenten van de extracellulaire matrix produceren, wat uiteindelijk de hartwand verhardt. De onderzoekers gebruikten twee modellen om dit proces te bestuderen: muizen die werden behandeld met het middel isoproterenol, dat betrouwbaar hartfibrose induceert, en humane cardiale fibroblasten die werden blootgesteld aan het signaalmolecuul TGF-β1, een bekende trigger van littekenvorming. In beide settings bepaalden ze hoe specifieke genen en eiwitten veranderden naarmate fibrose zich ontwikkelde.
Een schadelijke kettingreactie binnenin cellen
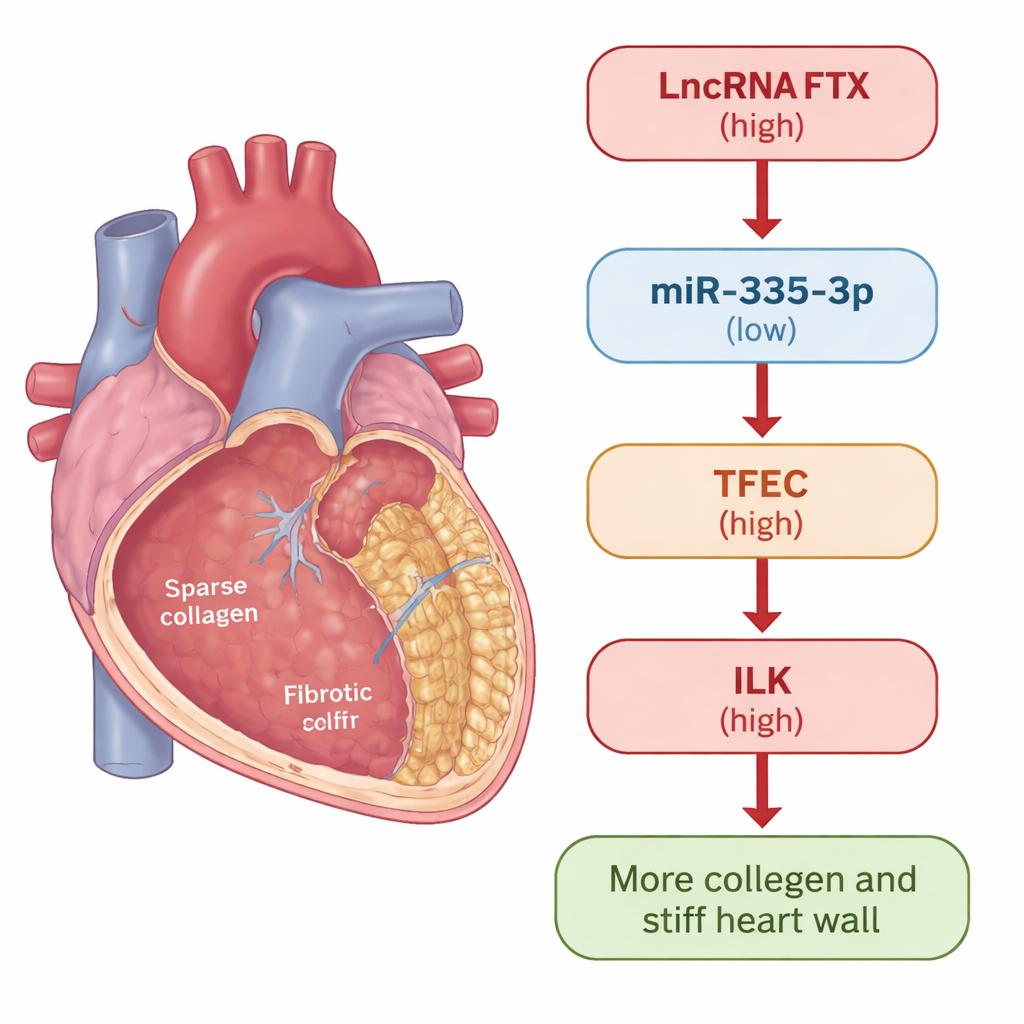
Het team richtte zich op een transcriptiefactor genaamd TFEC, een eiwit dat in de celkern zit en andere genen aanzet. Ze vonden dat TFEC, samen met een ander eiwit genaamd integrin-linked kinase (ILK), consistent verhoogd was wanneer fibroblasten naar een fibrotische, littekenvormende toestand werden gedrongen. Het uitschakelen van TFEC of ILK verlaagde markant klassieke fibrosemarkers zoals α-smooth muscle actin en collageen I en III, en dempte ook een groeireguleringspad (Akt/GSK3β en Hippo-signalisatie) dat bekend staat om weefselschade te bevorderen. Experimenten die DNA-binding in kaart brachten toonden aan dat TFEC direct bindt aan de promoter van het ILK-gen en de activiteit daarvan verhoogt, waarmee TFEC duidelijk stroomopwaarts van ILK in een pro-fibrotische signaalcascade wordt geplaatst.
RNA-schakelaars die de hoofdregelaar controleren
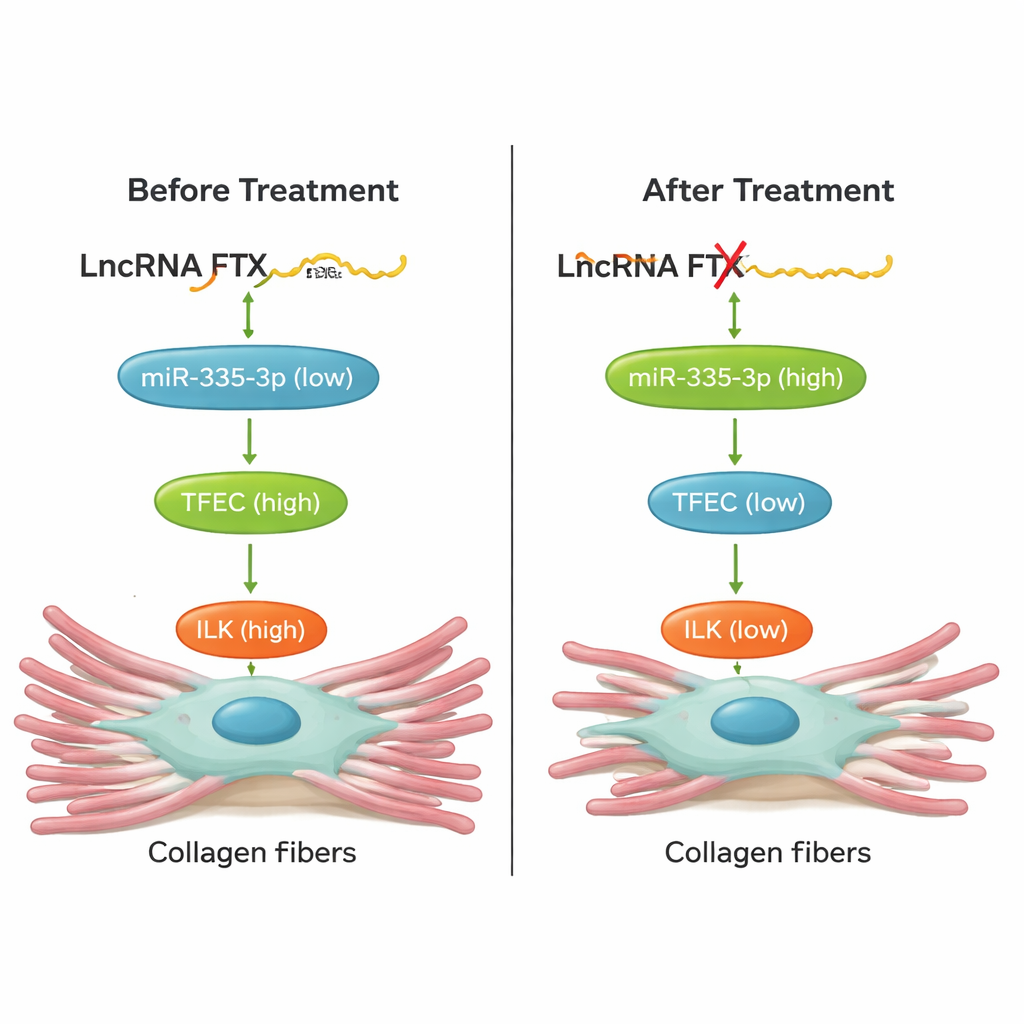
Om te begrijpen wat TFEC zelf reguleert, richtten de onderzoekers zich op niet-coderende RNA's—RNA-moleculen die geen eiwitten vormen maar fungeren als fijninstellers van genactiviteit. Ze identificeerden een klein RNA, miR‑335‑3p, dat verlaagd was in fibrotische harten en cellen. Het verhogen van miR‑335‑3p verlaagde TFEC, terwijl het blokkeren ervan TFEC verhoogde, en reporter-tests bevestigden dat miR‑335‑3p direct bindt aan TFEC-boodschappen om ze in toom te houden. Vervolgens vonden ze een lang niet-coderend RNA, genaamd FTX, dat verhoogd was bij fibrose en fysiek met miR‑335‑3p interageerde. FTX gedroeg zich als een moleculaire spons: het nam miR‑335‑3p op en verhinderde dat dit kleine RNA TFEC onder controle hield. Als gevolg daarvan stegen TFEC en ILK, en produceerden fibroblasten meer collageen dat littekenvorming bevordert.
Van celkweek naar levende harten
Cruciaal testte het team of het verstoren van deze keten harten in dieren daadwerkelijk kon beschermen. Bij muizen die aan isoproterenol werden blootgesteld, leidden het verlagen van TFEC, het knockdown van FTX in het hart met een AAV9-gentherapievector, of het verhogen van miR‑335‑3p met een chemisch gestabiliseerde "agomir" allemaal tot minder collageenophoping en lagere niveaus van fibrosemarkers in hartweefsel. Deze interventies verbeterden ook de hartfunctie: slagvolume en ejectiefractie gingen weer richting normaal, en schadelijke verhogingen van de hartfrequentie werden afgezwakt. Rescue-experimenten in cellen toonden dat het veranderen van een component van de FTX/miR‑335‑3p/TFEC/ILK-as de andere componenten voorspelbaar verschuift, wat bevestigt dat dit een strak verbonden pad is en geen losse correlatie.
Wat dit betekent voor toekomstige behandelingen
Voor niet-specialisten is de kernboodschap dat de auteurs een nieuwe "regelhendel" voor hartlittekenvorming hebben geïdentificeerd. Een lang RNA genaamd FTX haalt de remmen (miR‑335‑3p) weg van een hoofdschakelaar (TFEC), die vervolgens ILK en downstream pro-litteken signalen inschakelt, wat leidt tot overmatige collageenafzetting en verharding van het hart. Door FTX terug te dringen, miR‑335‑3p te herstellen of TFEC rechtstreeks te blokkeren, was het mogelijk om bij muizen littekenvorming te verminderen en de pompwerking te verbeteren. Hoewel meer werk nodig is om dit pad bij menselijke patiënten te bevestigen en veilige therapieën te ontwikkelen, biedt deze RNA-gebaseerde regulerende keten meerdere veelbelovende aanknopingspunten om in fibrose-gedreven hartfalen in te grijpen.
Bronvermelding: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Trefwoorden: myocardiale fibrose, hartfalen, niet-coderend RNA, cardiale fibroblasten, fibrose signalering