Clear Sky Science · nl
Machine learning interatomair potentieel voor de structurele eigenschappen van ijzeroxiden
Waarom roestige stenen ertoe doen
Ijzeroxiden – de mineralen die roest zijn kleur geven – ondersteunen stilletjes veel aspecten van het moderne leven. Ze zijn de belangrijkste bron van ijzer voor staal, essentiële bestanddelen in batterijen en zonnecellen, en helpen zelfs bij het zuiveren van vervuild water. Ondanks hun belang hebben we nog steeds moeite om te voorspellen hoe deze materialen zich onder reële omstandigheden gedragen, vooral op atomaire schaal. Dit artikel beschrijft hoe onderzoekers moderne kunstmatige intelligentie gebruikten om een snel, nauwkeurig digitaal model van één cruciaal ijzeroxide, hematiet, te bouwen, en zo de deur te openen naar betrouwbaardere virtuele experimenten — van ertsbewerking tot apparaten voor schone energie.
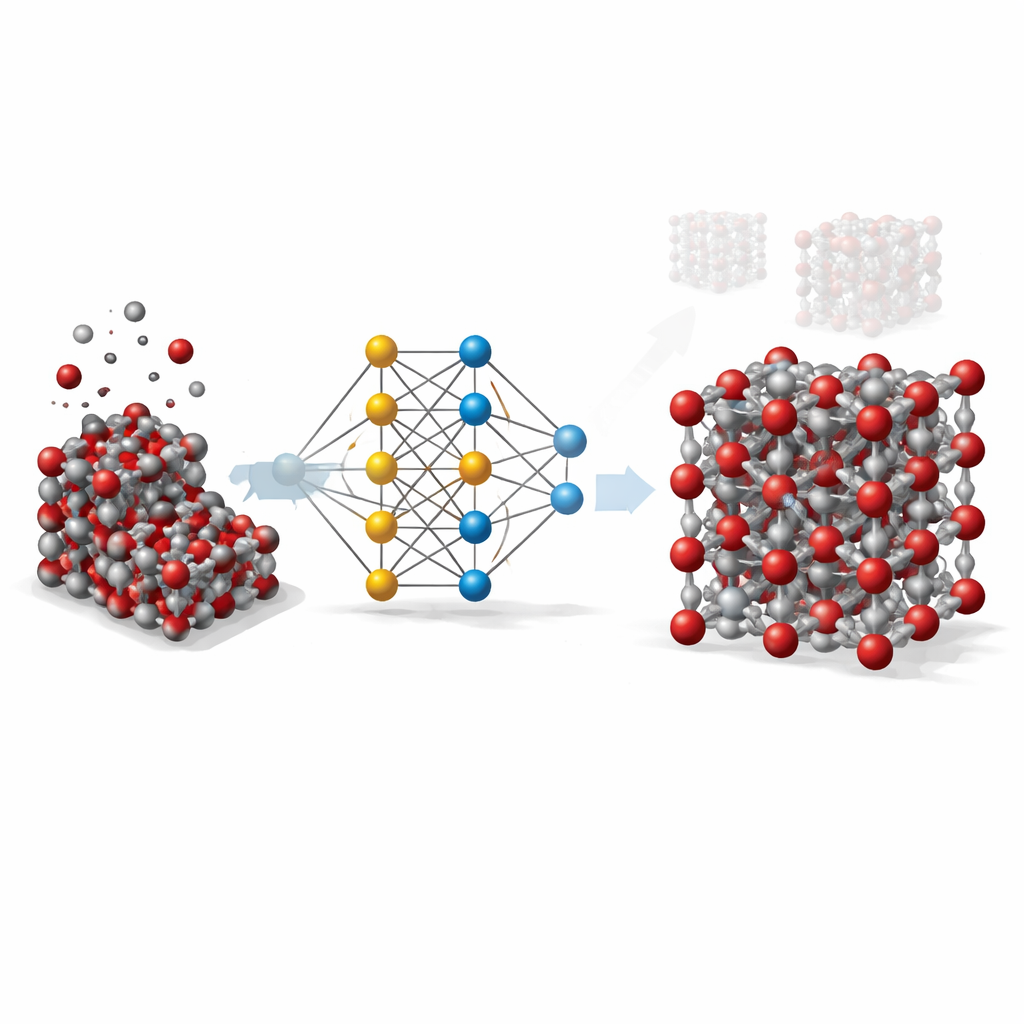
Van dure berekeningen naar slimme kortere routes
Om een vaste stof zoals hematiet in detail te begrijpen, vertrouwen wetenschappers idealiter op kwantummechanische methoden die volgen hoe elektronen en atomen op elkaar inwerken. Deze methoden zijn weliswaar zeer nauwkeurig, maar zo rekenintensief dat ze onpraktisch zijn voor het simuleren van grote stalen of lange tijdschalen. Klassieke modellen zijn daarentegen snel maar grof: ze leunen op eenvoudige formules die voor specifieke situaties zijn afgestemd en falen vaak wanneer temperatuur, druk of kristalvormen veranderen. Het hier gepresenteerde werk wil deze kloof overbruggen door machine learning te gebruiken om de nauwkeurigheid van kwantumberekeningen na te bootsen, terwijl de snelheid van traditionele modellen behouden blijft.
Een neuraal netwerk leren over atomen
Het team bouwde wat bekendstaat als een graph neural network-potentieel voor hematiet. In deze benadering wordt elk atoom behandeld als een knooppunt in een netwerk, en de bindingen en naburige atomen vormen de verbindingen tussen knooppunten. Om het netwerk te leren hoe atomen in hematiet elkaar aantrekken en afstoten, genereerden de onderzoekers eerst duizenden atomaire snapshots met standaardsimulaties over een breed bereik van temperaturen, drukken en kristalverstoringen, inclusief zowel bulkkristallen als blootgestelde oppervlakken. Vervolgens gebruikten ze een geavanceerde kwantummethode (DFT+U) om de energie, krachten en interne spanningen voor elk snapshot te berekenen, en trainden ze het neuraal netwerk om deze waarden zo nauwkeurig mogelijk te reproduceren.
Het model controleren aan de werkelijkheid
Nadat het getraind was, werd het nieuwe potentieel — genoemd Fe-MLIP — rigoureus getest. De auteurs vergeleken de voorspellingen voor basisstructurele grootheden zoals roosterafmetingen en hoe het kristal uitrekt onder spanning met zowel experimenten als meerdere veelgebruikte klassieke modellen. Fe-MLIP reproduceerde de bekende kristalstructuur van hematiet binnen enkele procenten en wist het elastische gedrag bijna even goed vast te leggen als directe kwantumberekeningen, waarbij het duidelijk beter presteerde dan andere krachtvelden voor veel eigenschappen. Het deed het ook goed op subtielere tests, zoals hoe het materiaal uitzet met temperatuur en hoe zijn atomen trillen — belangrijk voor warmtetransport en spectroscopie. Deze trillingfrequenties, die tijdens het trainen nooit expliciet werden getoond, lagen dichter bij de gemeten waarden dan die van concurrerende modellen.
Voorbij één mineraal reiken
De onderzoekers onderzochten vervolgens hoe ver dit op hematiet gebaseerde model kon worden uitgerekt. Ze pasten het toe op verwante ijzeroxiden — maghemit en magnetiet — die vergelijkbare atomaire bouwstenen delen maar verschillen in kristalordening en ijzerladingsstaten. Hoewel Fe-MLIP niet op deze fasen was getraind, leverde het redelijke waarden voor hun roosterafmetingen en stijfheid op, vaak gelijk aan of beter dan gespecialiseerde klassieke modellen. Het potentieel legde ook de relatieve stabiliteit van belangrijke kristaloppervlakken vast en zelfs trends in de energiekosten voor het creëren van atomaire vacanties, kenmerken die cruciaal zijn voor het begrijpen van corrosie, katalyse en batterijprestaties.
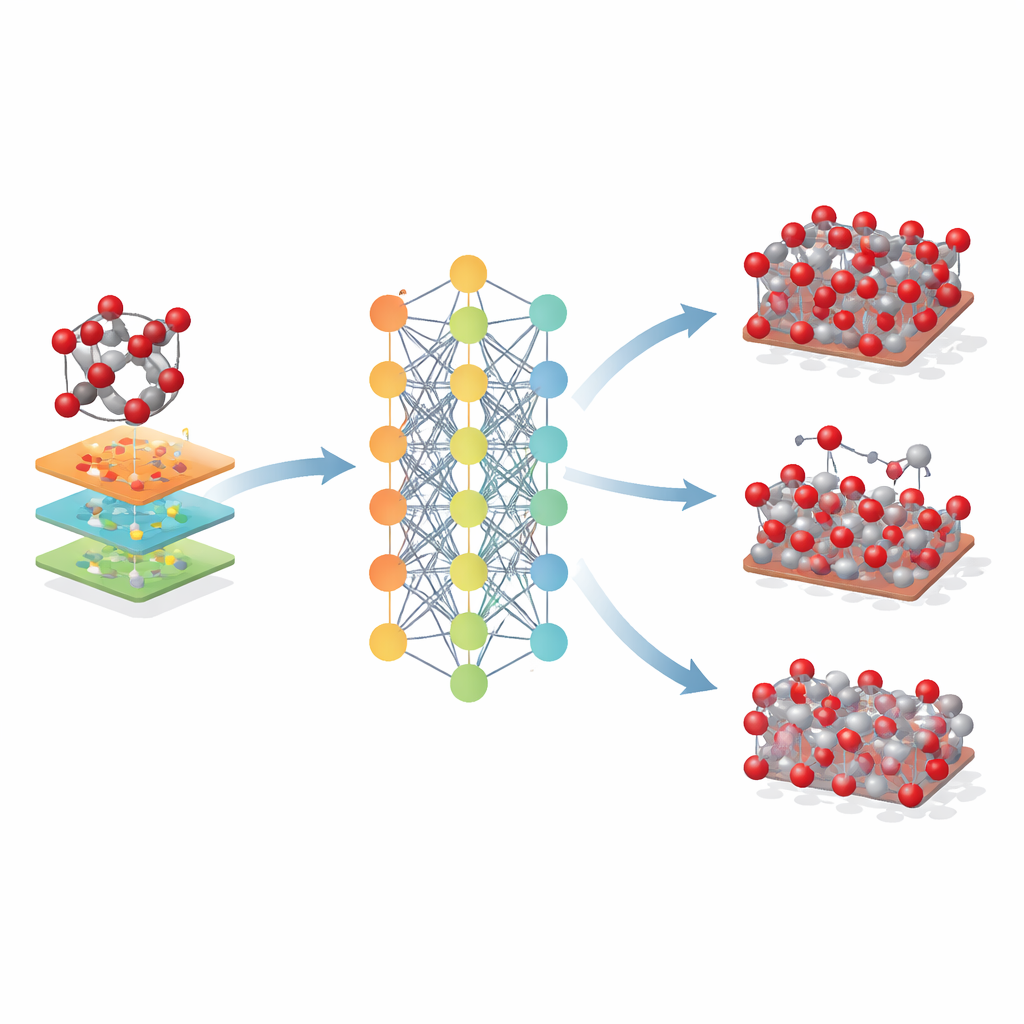
Wat dit betekent voor toekomstig materiaalontwerp
Voor niet-specialisten is de kernboodschap dat dit werk een krachtig nieuw "digitale tweeling" voor ijzeroxiden biedt. Het Fe-MLIP-model stelt onderzoekers in staat grote, lange simulaties van hematiet en aanverwante materialen uit te voeren met bijna kwantumniveau betrouwbaarheid, maar voor een fractie van de kosten. Hoewel het enkele beperkingen van de onderliggende kwantummethode erft en momenteel is gefocust op ijzer en zuurstof, maakt het al realistischere studies mogelijk van hoe deze mineralen reageren op spanning, warmte, oppervlakken en defecten. In praktische zin kan een dergelijk hulpmiddel het ontwerp van betere staalproductieprocessen, efficiëntere katalysatoren en batterijen, en verbeterde milieutechnologieën die afhankelijk zijn van ijzeroxiden versnellen — doordat wetenschappers ideeën op de computer kunnen testen voordat ze naar het laboratorium of de mijn gaan.
Bronvermelding: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Trefwoorden: hematiet, ijzeroxiden, machine learning potentieel, graph neural networks, moleculaire dynamica