Clear Sky Science · nl
Optimaliseren van kenmerkselectie in microarray‑gegevens bij kanker met een heap‑gestuurd evolutionair kader voor hoogdimensionale ruimten
Waarom het kiezen van de juiste genen ertoe doet
Moderne genetische tests bij kanker kunnen tegelijk tienduizenden genen meten, maar artsen beschikken vaak over gegevens van slechts enkele tientallen patiënten. Verborgen in deze enorme “genjungle” bevindt zich een veel kleiner aantal signalen dat daadwerkelijk het ene type kanker van het andere of een tumor van gezond weefsel onderscheidt. Dit artikel presenteert een nieuwe slimme zoekmethode om automatisch die sleutelgenen te selecteren, met als doel computerondersteunde kankerdetectie nauwkeuriger, sneller en beter interpreteerbaar te maken.
Te veel signalen, te weinig gegevens
Microarray‑experimenten en vergelijkbare technieken stellen onderzoekers in staat de activiteit van duizenden genen per patiëntmonster te meten. Het aantal monsters is echter meestal zeer beperkt, soms minder dan honderd. Veel van deze genmetingen zijn ruisig, redundant of irrelevant voor de betreffende ziekte. Alles bewaren kan leeralgoritmen overweldigen, berekeningen vertragen en misleidende modellen opleveren die zich vastklampen aan toevalligheden in plaats van aan echte biologische signalen. Het proces om dit terug te brengen tot een bruikbare subset heet “kenmerkselectie” en is cruciaal om betrouwbare voorspellingen uit hoogdimensionale medische data te halen.
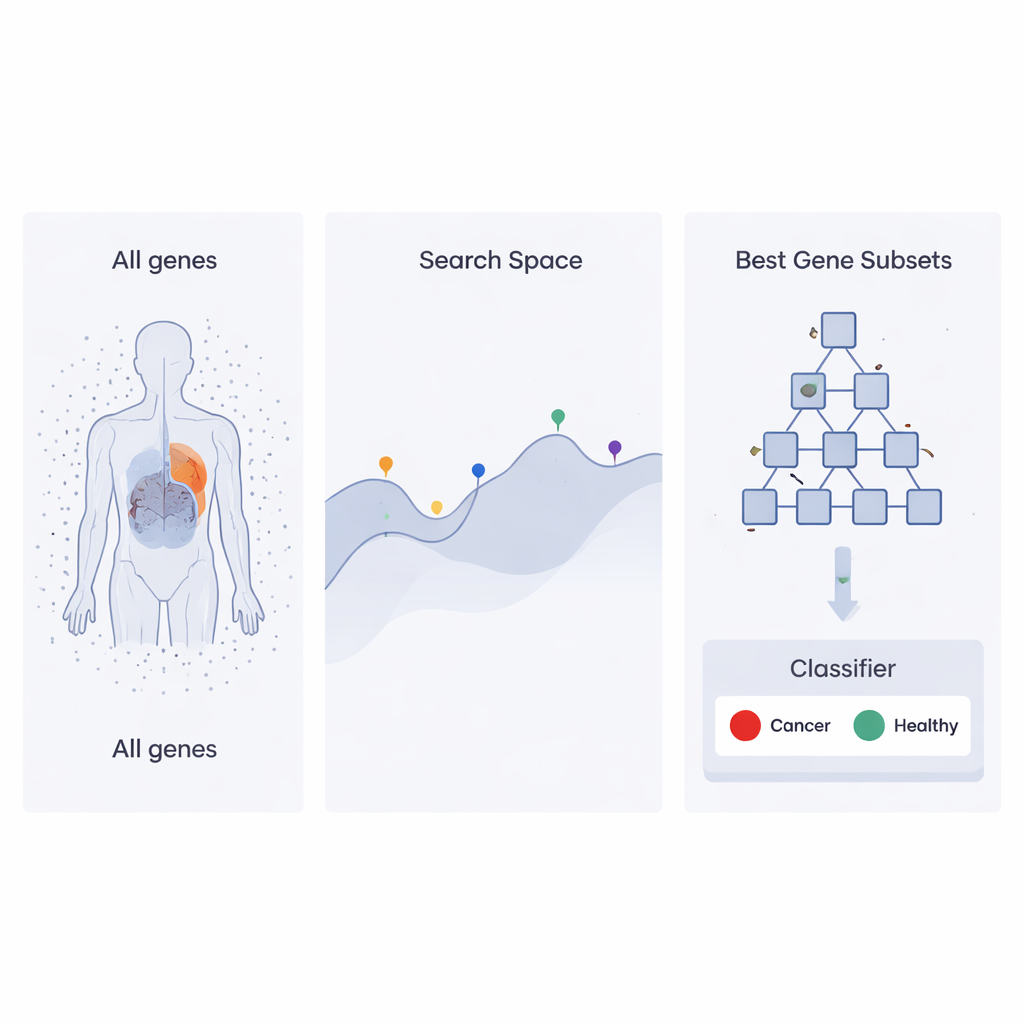
Een zoekstrategie geïnspireerd op bedrijfshiërarchieën
De auteurs bouwen voort op een recente optimalisatiebenadering genaamd Heap‑Based Optimizer (HBO), die ideeën ontleent aan hoe werknemers in een bedrijf georganiseerd zijn. Stel je elke mogelijke set genen voor als een “werknemer” waarvan de prestaties worden beoordeeld op basis van hoe goed die set een classifier helpt kankermonsters van gezonde monsters te onderscheiden. Deze werknemers worden in een hiërarchie geplaatst, als een bedrijfsladder, met behulp van een computerstructuur die een heap wordt genoemd. Hoog presterende genensets zitten dichtbij de top, zwakkere sets lager. Gedurende vele ronden passen lager geplaatste werknemers hun keuzes aan door te kopiëren en licht te variëren wat hun bazen en collega’s doen, waardoor de hele organisatie geleidelijk betere oplossingen gaat vinden.
Ruwe gengegevens omzetten in scherpere patronen
Om de zoekactie effectiever te maken, vertrouwen de auteurs niet alleen op ruwe genwaarden. Ze herschikken eerst de microarraygegevens naar een afbeelding‑achtige vorm en passen een techniek toe genaamd Histogram of Oriented Gradients (HOG), veelgebruikt in computerzicht. HOG legt vast hoe expressieniveaus over genen veranderen en benadrukt lokale patronen in plaats van geïsoleerde metingen. Deze patroongebaseerde kenmerken worden vervolgens gecombineerd met de oorspronkelijke geninformatie. Een eenvoudige classifier, k‑Nearest Neighbors (KNN), fungeert als de “rechter”, en scoort elke kandidaat‑genset op hoe accuraat die nieuwe monsters labelt, terwijl ook kleinere, compactere sets worden beloond.
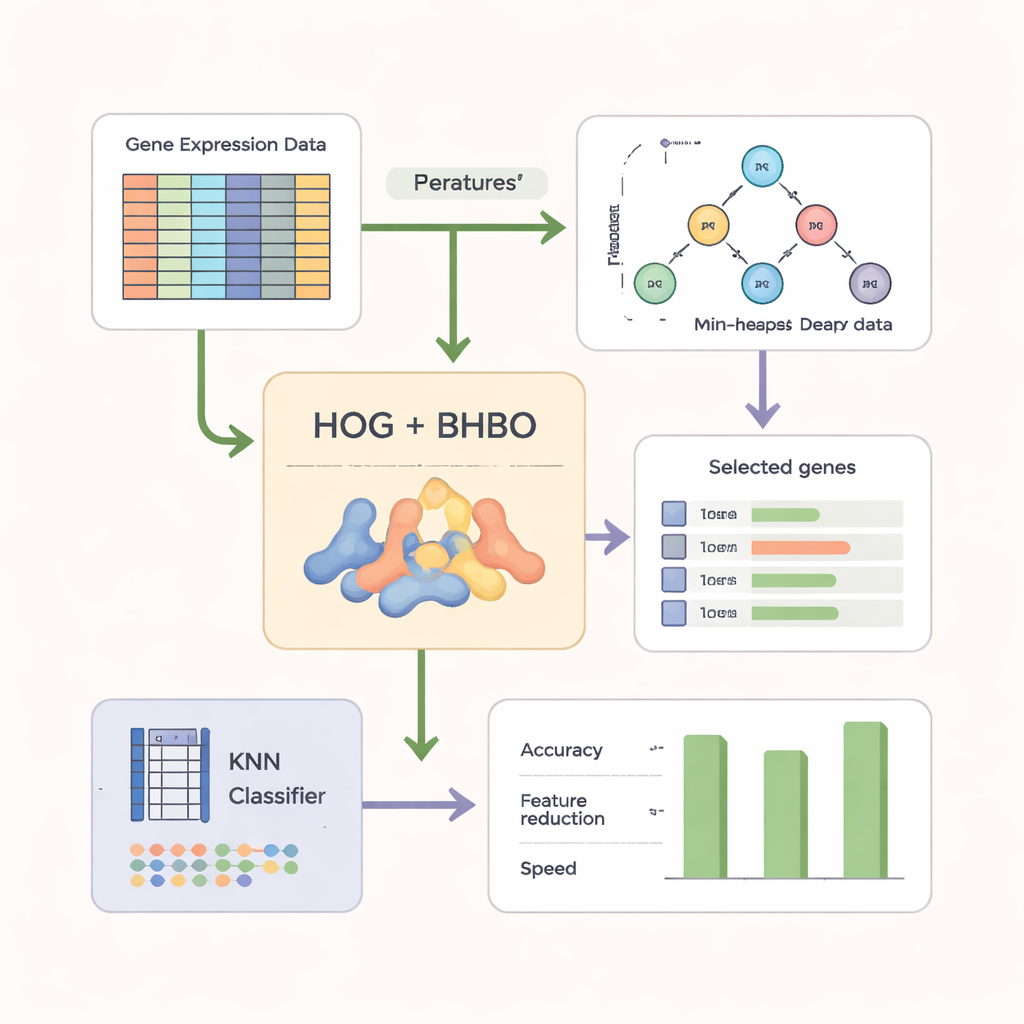
Testen op meerdere kankerdatasets
De onderzoekers evalueerden hun binaire versie van de Heap‑Based Optimizer (BHBO) op negen openbare kanker‑microarraydatasets, waaronder hersentumoren, leukemieën, prostaatkanker en samengestelde tumoren met veel subtypen. Elke dataset bevatte duizenden tot meer dan vijftienduizend gemeten genen, maar relatief weinig patiëntmonsters. Voor elke dataset werd BHBO herhaaldelijk uitgevoerd en vergeleken met zeven bekende zoekmethoden, zoals genetische algoritmen en particle swarm optimization. Het team keek niet alleen naar nauwkeurigheid, maar ook naar hoeveel genen behouden werden, hoe snel de zoekopdracht convergeerde en hoe stabiel de resultaten waren wanneer de data verstoord werden door gesimuleerde ruis, batch‑effecten en foutieve labels.
Wat de nieuwe methode bereikte
Over de negen datasets behaalde de heap‑gestuurde aanpak een gemiddelde classificatienauwkeurigheid van ongeveer 95 procent, terwijl het aantal genen met meer dan 85 procent werd teruggebracht. Het overtrof concurrerende methoden duidelijk op meerdere datasets en liet snellere convergentie zien, wat betekent dat het in minder zoekstappen tot goede gensets kwam. Zelfs wanneer de auteurs de data opzettelijk corrumpeerden door ruis toe te voegen of sommige sample‑labels om te draaien, daalde de prestatie slechts licht en bleef beter dan die van de alternatieven. Statistische toetsen bevestigden dat deze verbeteringen waarschijnlijk niet door toeval verklaard konden worden.
Wat dit betekent voor toekomstige kankerdiagnostiek
In praktische termen laat dit werk zien dat een zorgvuldig ontworpen zoekstrategie door enorme genetische datasets kan ziften en kleine, informatie‑rijke panels van genen kan onthullen die nog steeds tumoren zeer goed classificeren. Voor clinici en onderzoekers zijn zulke compacte gensets makkelijker biologisch te valideren, goedkoper te meten in vervolgtests en beter geschikt voor integratie in beslissingsondersteunende tools. Hoewel de methode niet direct nieuwe geneesmiddelen of paden ontdekt, richt ze wel de aandacht op veelbelovende genetische markers en helpt ze andere studies te focussen op de meest informatieve signalen die verborgen liggen in hoogdimensionale kankergegevens.
Bronvermelding: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Trefwoorden: kanker microarray, kenmerkselectie, metaheuristieke optimalisatie, gene biomarkers, medische data‑mining