Clear Sky Science · nl
Een DNABERT-gebaseerd deep learning-framework voor het voorspellen van bindingsplaatsen van transcriptiefactoren
Waarom het voorspellen van DNA-besturingsschakelaars ertoe doet
Elke cel in je lichaam draagt in wezen hetzelfde DNA, maar hersencellen, levercellen en immuuncellen gedragen zich heel verschillend. Een belangrijke reden is dat speciale eiwitten, transcriptiefactoren genoemd, als moleculaire schakelaars werken en genen aan- of uitzetten door zich te hechten aan korte DNA-stukjes die bindingsplaatsen worden genoemd. Deze aanlegpunten experimenteel in het hele genoom vinden is traag en duur. Deze studie introduceert TFBS-Finder, een nieuw kunstmatig-intelligentie-model dat ruwe DNA-letters kan lezen en nauwkeuriger kan voorspellen waar transcriptiefactoren binden, wat onderzoek naar genregulatie en ziekte kan versnellen.
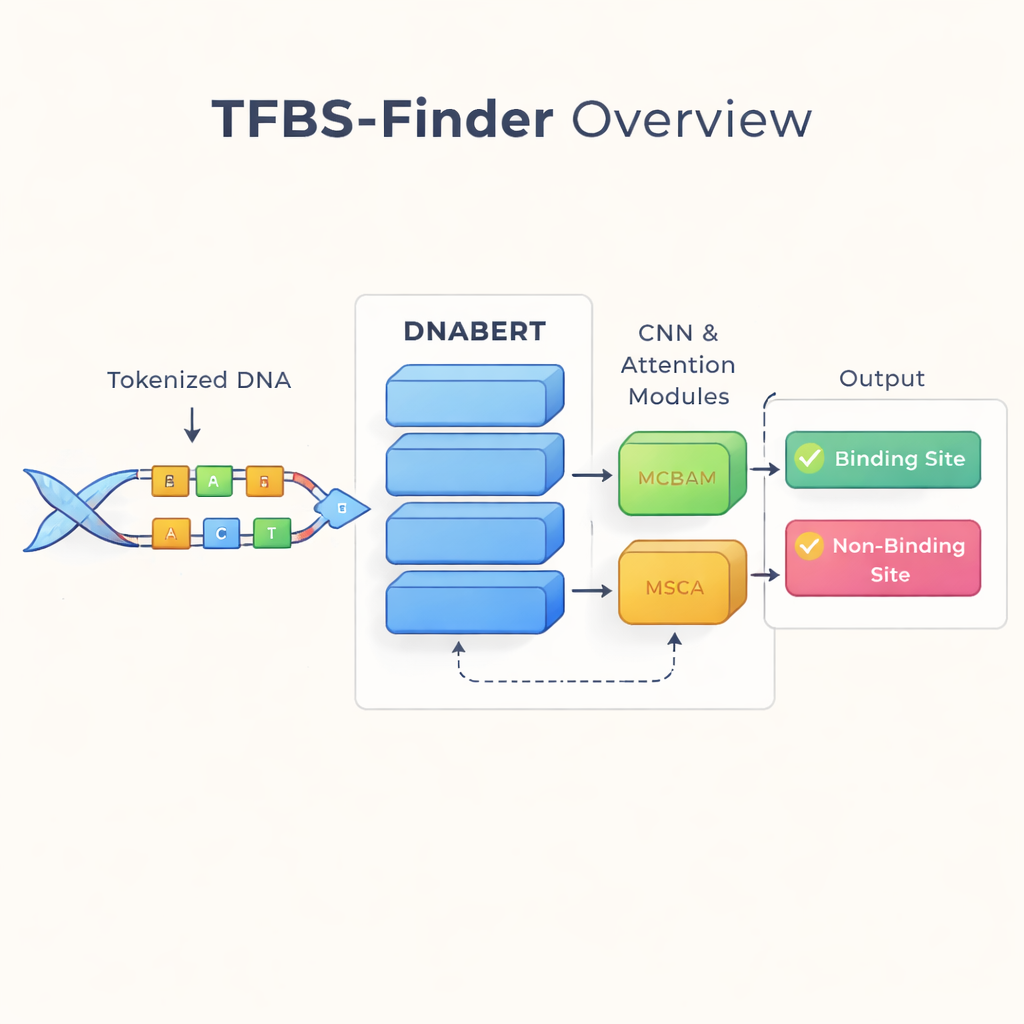
DNA lezen als een taal
De auteurs bouwen voort op een idee dat de taalkundige technologie heeft getransformeerd: behandel DNA alsof het tekst is. Ze gebruiken DNABERT, een versie van het BERT-taalmodel die is bijgetraind op menselijk DNA in plaats van woorden. DNABERT kijkt niet alleen naar enkele letters; het splitst DNA op in overlappende korte “woorden” van vijf letters en leert hoe die stukjes doorgaans samen voorkomen. Dit stelt het model in staat langeafstandscontext te vangen, bijvoorbeeld hoe patronen aan het ene uiteinde van een sequentie zich verhouden tot patronen ver weg, vergelijkbaar met het begrijpen van de betekenis van een zin in plaats van geïsoleerde woorden.
Lokale patronen vinden met gerichte aandacht
Hoewel DNABERT goed is in het vatten van globale context, hangt het binden van transcriptiefactoren vaak af van zeer korte, precieze motieven—lokale patronen in het DNA. TFBS-Finder voegt daarom meerdere extra componenten toe bovenop DNABERT. Een convolutioneel neuraal netwerk (CNN) kamt door de sequentie-embeddings om terugkerende lokale vormen te benadrukken, vergelijkbaar met hoe beeldsoftware randen en hoeken detecteert. Twee attentie-modules, MCBAM en MSCA genoemd, fungeren vervolgens als instelbare spots die de meest informatieve kenmerken versterken en ruis onderdrukken. Samen balanceren deze blokken het totaalbeeld met fijnmazige details om te beslissen of een DNA-segment een echte bindingsplaats bevat.
Aantonen dat elk onderdeel echt helpt
Om te testen of al deze componenten nodig zijn, voerde het team uitgebreide "ablatieve" experimenten uit, waarbij ze systematisch modules verwijderden of herordenden en het systeem opnieuw trainden op 165 benchmarkdatasets die 29 transcriptiefactoren over 32 celtypen beslaan. Met standaardmaten voor voorspellingskwaliteit kwam het volledige TFBS-Finder-model consequent als beste uit de bus. Eenvoudigere versies die alleen op DNABERT vertrouwden of een van de attentie-modules weglieten, verloren duidelijk aan nauwkeurigheid. Statistische toetsen bevestigden dat deze prestatieverliezen geen toeval waren, wat aantoont dat de combinatie van globaal sequentiebegrip en zorgvuldig ontworpen aandacht voor lokale patronen cruciaal is.
Werken over celtypen heen en oudere tools overtreffen
Een belangrijke vraag is of een model dat in één biologische context is getraind, kan generaliseren naar een andere. De auteurs concentreerden zich op een goed bestudeerde transcriptiefactor, CTCF, en trainden TFBS-Finder op gegevens uit één cellijn en testten het vervolgens op andere. In alle combinaties behaalde het model hoge scores, wat suggereert dat het kernkenmerken van CTCF-binding vastlegt die in verschillende weefsels gedeeld worden. In vergelijking met negen toonaangevende methoden, waaronder eerdere deep learning- en BERT-gebaseerde modellen, toonde TFBS-Finder hogere gemiddelde nauwkeurigheid en produceerde het betrouwbaardere rangschikkingen van bindingsplaatsen. Het liep ook iets sneller en gebruikte minder geheugen dan het meest vergelijkbare eerdere model, wat aangeeft dat betere prestaties niet meer rekenkracht vereisten.
Zien wat het model heeft geleerd
Complexe AI-systemen worden vaak bekritiseerd als "black boxes." Hier probeerden de onderzoekers die doos te openen door te visualiseren welke DNA-posities de beslissingen van TFBS-Finder het meest beïnvloedden. Voor twee transcriptiefactoren met goed bekende bindingsmotieven, CEBPB en GATA3, genereerden ze belangscores langs de sequentie en clusteren ze de sterkste signalen tot consensuspatronen. Deze gereconstrueerde motieven kwamen nauw overeen met referentiemotieven uit gevestigde databases, en de voorspelde bindingsregio’s overlapten met onafhankelijk gedetecteerde motiefinstanties. Dit suggereert dat TFBS-Finder niet simpelweg voorbeelden uit het hoofd leert maar biologisch betekenisvolle regels heeft geleerd over hoe transcriptiefactoren DNA herkennen.
Wat dit betekent voor genetica en geneeskunde
TFBS-Finder biedt een nauwkeurigere en beter interpreteerbare manier om de besturingsschakelaars in ons DNA in kaart te brengen. Door vast te stellen waar transcriptiefactoren waarschijnlijk binden, kan het onderzoekers helpen genregulerende netwerken in kaart te brengen, te prioriteren welke genetische varianten cruciale besturingsplaatsen zouden kunnen verstoren, en gerichtere experimenten te ontwerpen. Hoewel het huidige werk geshuffelde sequenties als kunstmatige negatieven gebruikt en zich alleen op DNA-letters richt, zijn de auteurs van plan structurele informatie over DNA-vorm toe te voegen en realistischere achtergrondsequenties te verkennen. Naarmate deze modellen verbeteren, zouden ze krachtige hulpmiddelen kunnen worden om te begrijpen hoe veranderingen in niet-coderend DNA bijdragen aan ontwikkeling, evolutie en ziektarisico.
Bronvermelding: Dutta, P., Ghosh, N. & Santoni, D. A DNABERT based deep learning framework for predicting transcription factor binding sites. Sci Rep 16, 7018 (2026). https://doi.org/10.1038/s41598-026-37483-1
Trefwoorden: bindingsplaatsen van transcriptiefactoren, deep learning, DNABERT, genregulatie, genomics