Clear Sky Science · nl
Aanpasbare fuzzy-cluster-gestuurde eenvoudige, snelle en efficiënte kenmerkselectie voor hoog-dimensionale en sterk ongebalanceerde binaire bioinformatica microarraygegevens
Waarom dit belangrijk is voor genonderzoek
Moderne genexpressietests kunnen tienduizenden genen in een enkel patiëntmonster meten. Die datastroom belooft vroegere kankerdiagnoses en betere behandelkeuzes, maar veroorzaakt ook een probleem: de meeste van die genen zijn ruis, redundant of vooral gekoppeld aan veelvoorkomende gevallen, niet aan de zeldzame en gevaarlijke. Dit artikel presenteert een nieuwe manier om enorme genexpressiedatasets te doorzoeken zodat computers betrouwbaar patiënten in een kleine, moeilijk te detecteren minderheidsgroep kunnen herkennen met slechts een klein, zorgvuldig gekozen genpanel.
De uitdaging van te veel, te vergelijkbare genen
Microarrayexperimenten volgen vaak duizenden genactiviteitsniveaus voor slechts een paar honderd patiënten. Gewoonlijk overtreft de ene klasse (zoals een veelvoorkomend kankersubtype) de andere sterk, wat leidt tot sterk ongebalanceerde data. In deze situatie gedragen veel genen zich op zeer vergelijkbare manieren en kunnen de patronen van meerderheid- en minderheidspatiënten overlappen. Standaard leermethoden neigen ertoe zich vast te klampen aan de meerderheidsklasse en raken in de war door redundante genen, wat overfitting en slechte detectie van zeldzame subtypen veroorzaakt. Traditionele dimensionaliteitsreductiemethoden gooien ofwel de interpretatie overboord door nieuwe gemengde kenmerken te bouwen, of selecteren genen zonder goed te bekijken hoezeer ze een classifier helpen de minderheidsgroepen te herkennen.
Een nieuw stappenplan voor slimmer genselectie
De auteurs introduceren AFCG‑SFE, een adaptief kenmerkselectiemodel dat specifiek is ontworpen voor hoog-dimensionale, ongebalanceerde genexpressiegegevens. De methode begint met een eenvoudige “single-agent” zoekmethode die genen aan- of uitzet en test hoe goed ze classificatie ondersteunen, maar verrijkt dit met meerdere data-gedreven stappen. Eerst groepeert het genen op basis van overeenkomend gedrag, en laat het genen tot meer dan één groep behoren om de biologische realiteit te weerspiegelen dat een gen bij meerdere paden betrokken kan zijn. Binnen elke groep rangschikt het de genen op hoe informatief ze zijn voor het ziekte-label en behoudt slechts een paar sleutelvertegenwoordigers, waardoor redundantie scherp wordt teruggedrongen nog voordat de hoofdzoektocht begint. 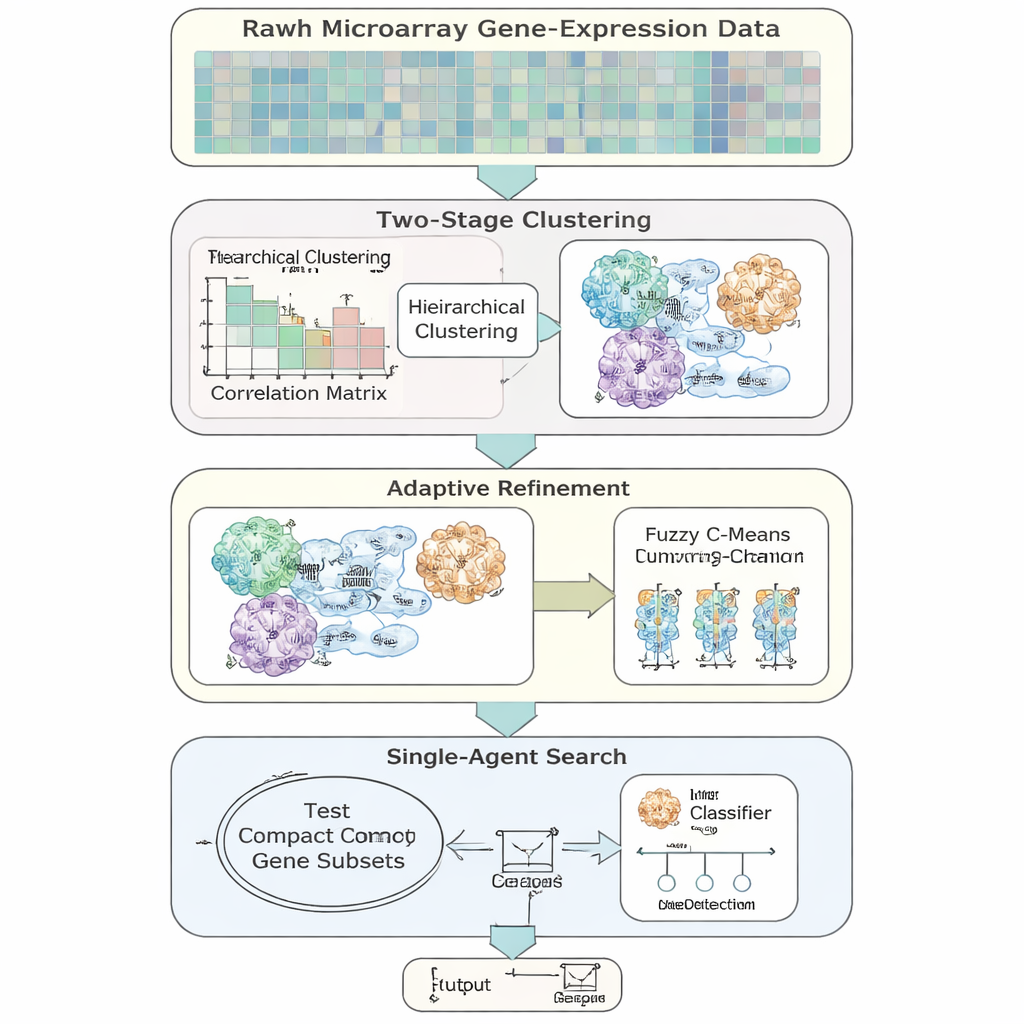
De computer laten letten op zeldzame patiënten
In plaats van zich te richten op gewone nauwkeurigheid, gebruikt AFCG‑SFE een fitnessscore die metriek benadrukt die geschikt is voor scheve gegevens, waaronder de balans tussen het correct identificeren van minderheids- en meerderheidsgevallen en prestaties over alle beslissingsdrempels. De fitnessfunctie bevat ook straffen voor het selecteren van te veel genen of veel genen uit dezelfde cluster, en een beloning voor genen die sterke afhankelijkheid met het ziekte-label vertonen. Belangrijk is dat de sterkte van deze straffen en beloningen automatisch wordt ingesteld vanuit eigenschappen van de dataset, zoals hoeveel genen er per patiënt zijn en hoeveel de klassen overlappen, in plaats van handmatige afstemming. Dit maakt de methode robuuster en gemakkelijker over te dragen tussen studies.
Aanpassen aan de moeilijkheid van het probleem
Een kernidee is dat het algoritme niet altijd moet mikken op de kleinst mogelijke genenset. Wanneer de twee klassen zeer moeilijk te scheiden of sterk overlappend zijn, verhoogt de methode automatisch een ondergrens voor hoeveel genen behouden moeten blijven, zodat zeldzame maar belangrijke signalen niet worden weggegooid. Naarmate de zoekprocedure vordert, versmalt AFCG‑SFE geleidelijk een per-cluster limiet op hoeveel genen uit elke groep kunnen overleven, terwijl het deze minimumgrens respecteert. Het resultaat is een compact, divers genpanel dat de structuur van de data vastlegt zonder te worden gedomineerd door één redundante patroon. 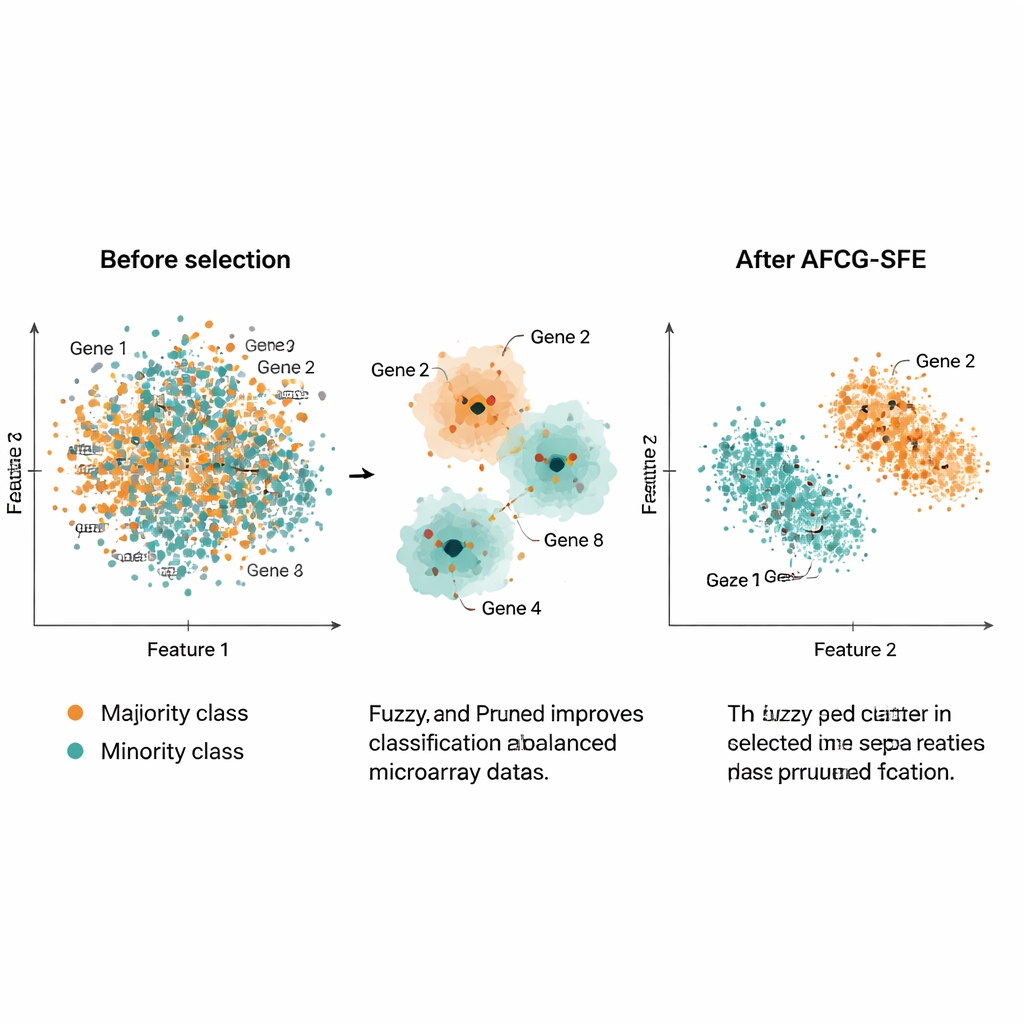
Wat de experimenten aantonen
De auteurs testten AFCG‑SFE op 20 openbare kanker-microarraydatasets, elk met duizenden genen maar slechts ongeveer 100–200 monsters en sterke klasse-onbalans. Ze vergeleken hun methode met meerdere evolutionaire zoekbasissen, eenvoudige filters en embedded benaderingen die kenmerkselectie in de classifier inbouwen. Over een reeks maatstaven — inclusief F‑maat, gebalanceerde nauwkeurigheid, oppervlakte onder de ROC‑curve en een maat voor overfitting — was AFCG‑SFE op alle datasets de beste of gedeeld beste. Het selecteerde doorgaans minder dan 25 genen (vaak slechts 6–8), waarbij meer dan 99% van de oorspronkelijke kenmerken werd verwijderd terwijl de classificatieprestaties werden verbeterd of behouden. Het verminderde ook een complexiteitsindex die vastlegt hoeveel de klassen in de kenmerkenruimte overlappen, wat wijst op een duidelijkere scheiding na selectie.
Conclusie voor niet‑experts
In praktische zin biedt dit werk een manier om enorme, ruizige genexpressieprofielen terug te brengen tot zeer kleine sets informatieve genen die computers toch in staat stellen zeldzame patiëntsubgroepen nauwkeurig te herkennen. Door vergelijkbare genen intelligent te groeperen, genen te belonen die echt de ziekte volgen, en expliciet te beschermen tegen bias naar de meerderheidsklasse, levert AFCG‑SFE zowel betere voorspelling als veel eenvoudigere genpanels. Die combinatie kan onderzoekers helpen te focussen op potentiële biomarkers, beter interpreteerbare diagnostische tests te ontwerpen en uiteindelijk te verbeteren hoe precisiegeneeskunde omgaat met echte, imperfecte biologische data.
Bronvermelding: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Trefwoorden: genexpressie, kenmerkselectie, ongebalanceerde gegevens, microarray, kankersubtypen