Clear Sky Science · nl
rhinotypeR maakt reproduceerbare toewijzing van rhinovirus-genotypes mogelijk op basis van VP4/2-sequenties
Waarom piepkleine verkoudheidsvirussen nog steeds van belang zijn
De meesten van ons zien de verkoudheid als een hinderlijke kwaal in plaats van een ernstige bedreiging. Toch worden de virussen die veel verkoudheden veroorzaken — humane rhinovirussen — ook in verband gebracht met ernstige longinfecties, astma-aanvallen en opvlammingen van chronische longaandoeningen. Om te volgen hoe deze virussen zich ontwikkelen en verspreiden, moeten onderzoekers ze in nauwkeurige genetische “types” indelen, vergelijkbaar met het toewijzen van streepjescodes aan producten. Dit artikel introduceert rhinotypeR, een gratis, open-source softwarepakket dat deze genetische labeling nauwkeuriger, consistenter en gemakkelijker reproduceerbaar maakt, en zo volksgezondheidsinstanties helpt een helderder beeld te houden van een vaak over het hoofd gezien familie van luchtwegvirussen.
De verborgen variatie in verkoudheden
Humane rhinovirussen komen buitengewoon veel voor; ze worden aangetroffen in tot wel 60% van de monsters van mensen met plotselinge luchtwegaandoeningen. Het zijn niet één, maar drie hoofdgroepen, genaamd A, B en C, met minstens 169 erkende genetische types. Verschillende types gedragen zich anders: sommige worden vaker gekoppeld aan ernstige infecties bij kinderen en aan astma‑opvlammingen, terwijl andere minder vaak voorkomen bij ernstige ziekte. Omdat deze types onafhankelijk evolueren en verschillende oppervlaktestructuren hebben, hebben wetenschappers betrouwbare methoden nodig om ze te onderscheiden als ze willen volgen hoe uitbraken zich door scholen, huishoudens en gemeenschappen verplaatsen.
Van versnipperde tools naar één duidelijke route
Tot nu toe was het toewijzen van een rhinovirus-type op basis van zijn genetische code een lappendeken van werkwijzen. Onderzoekers concentreerden zich doorgaans op een korte regio van het viraal genoom, de VP4/2-regio, alignen die met bekende referentiestammen, maten de onderlinge verschillen tussen sequenties en pasten vervolgens grenswaarden toe om te beslissen tot welk type elk monster behoorde. Die stappen werden echter uitgevoerd met een mix van softwareprogramma’s, handmatige bewerkingen en persoonlijke afwegingen. Dat maakte het moeilijk om verschillende studies met elkaar te vergelijken of te reproduceren, zelfs wanneer ze vergelijkbare gegevens gebruikten. rhinotypeR is specifiek gemaakt om dit meerstaps, foutgevoelige proces om te zetten in één gescripte workflow die iedereen kan uitvoeren en delen.
Wat de nieuwe software precies doet
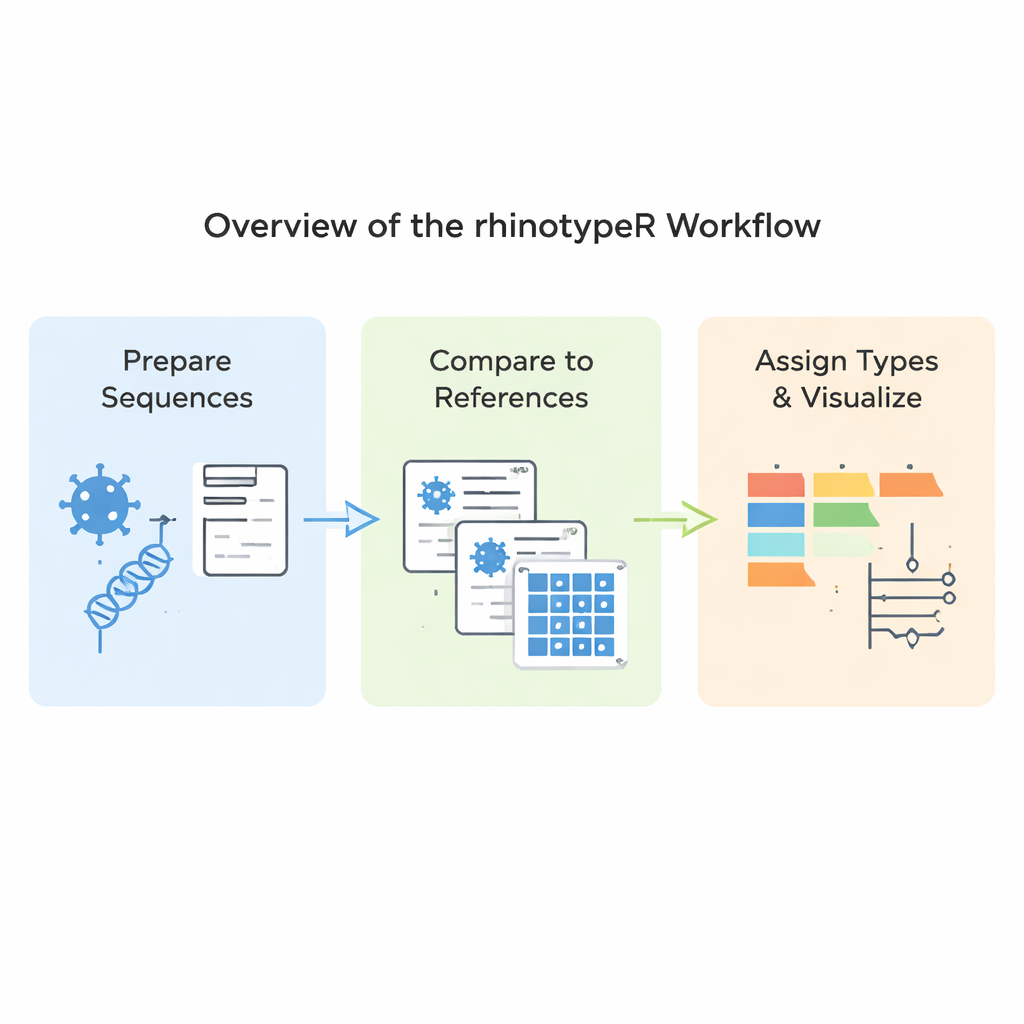
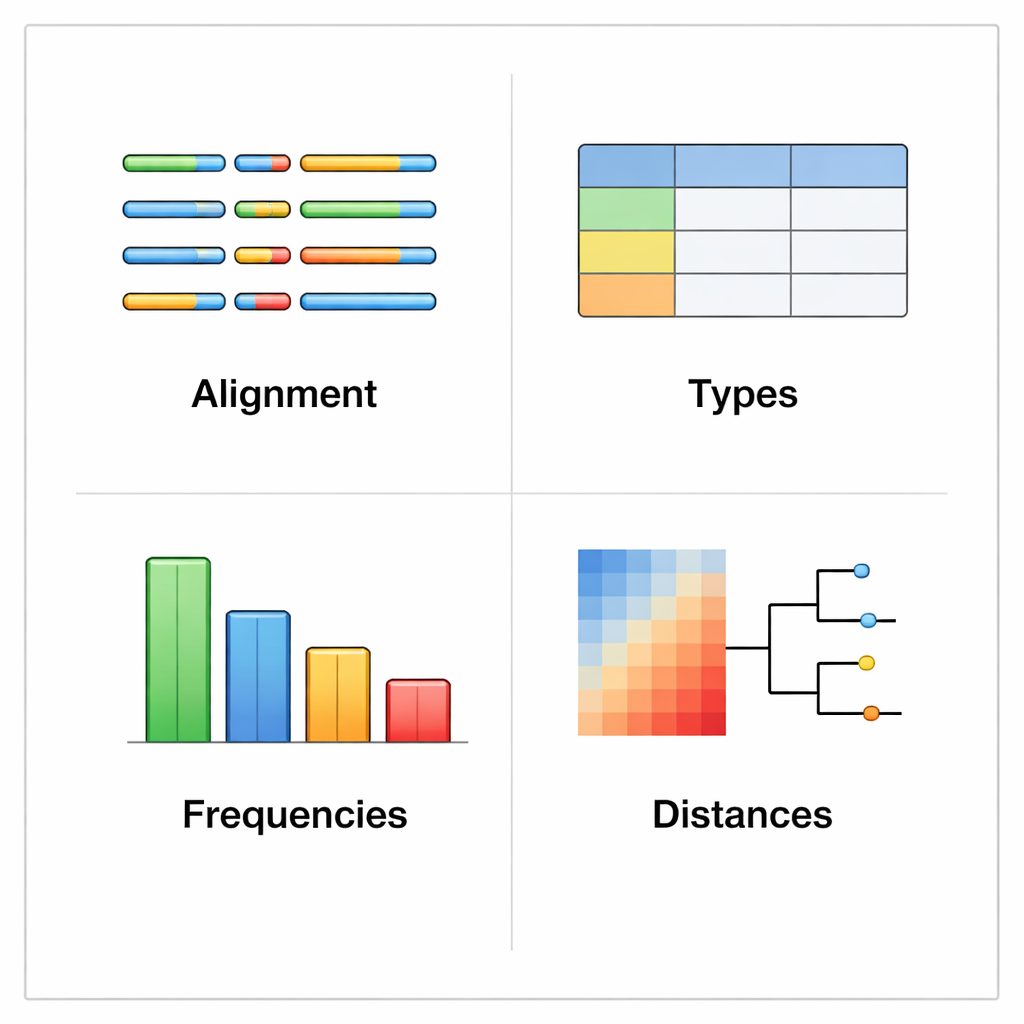
rhinotypeR draait binnen de veelgebruikte R- en Bioconductor-omgeving voor data-analyse. Het neemt een verzameling rhinovirus VP4/2-sequenties en voert ze langs drie hoofdstadia: het voorbereiden en alignen van de sequenties, het berekenen van de onderlinge afstanden tot een gecureerde set referentietypes, en vervolgens het toewijzen van elk monster aan het dichtstbijzijnde bekende type of het markeren als “niet-toegewezen” als het te verschillend is. Dezelfde tool kan visuele output produceren, waaronder kleurgecodeerde kaarten van genetische verschillen, eenvoudige stambomen en grafieken die laten zien hoe vaak elk type in een dataset voorkomt. Gebruikers kunnen hun data met externe programma’s alignen als ze dat liever willen, of rhinotypeR het hele proces binnen R laten afhandelen voor maximale reproduceerbaarheid.
Het gereedschap op de proef gesteld
Om te controleren of rhinotypeR betrouwbare resultaten levert, vergeleken de auteurs de afstandsmetingen met die van twee gevestigde programma’s, ape en MEGA X, met dezelfde invoerbestanden en modellen. De resultaten kwamen bijna volledig overeen; eventuele kleine verschillen waren te wijten aan normale afronding in rekenkundige bewerkingen, niet aan echte methodologische verschillen. Het team voerde rhinotypeR vervolgens uit op een grote verzameling van meer dan 2.300 rhinovirussequenties uit meerdere eerdere studies, die meer dan 90% van de bekende types besloegen. In ongeveer vier van de vijf gevallen kwam het nieuwe hulpmiddel exact overeen met eerdere type-aanduidingen. De meeste onenigheden deden zich voor rond de vooraf afgesproken grenswaarden die worden gebruikt om het ene type van het andere te scheiden — precies daar waar grensgevallen te verwachten zijn. Belangrijk is dat monsters die niet betrouwbaar aan een bekend type konden worden toegewezen, geen aanwijzingen vertoonden dat het simpelweg om monsters van slechte kwaliteit of met een laag virale lading ging, wat suggereert dat ze echte virale diversiteit kunnen weerspiegelen.
Waarom dit van belang is voor de volksgezondheid
Voor niet‑specialisten is de kernboodschap dat rhinotypeR niet opnieuw uitvindt hoe wetenschappers verkoudheidsvirussen classificeren; het maakt dat proces juist helderder, transparanter en gemakkelijker te reproduceren. Door alignering, afstandsberekeningen en type-toewijzing te bundelen in één open-source pakket — samen met duidelijke visuele samenvattingen — helpt het onderzoekers en surveillancesystemen om duizenden monsters op een consistente manier te verwerken. Die consistentie verbetert ons vermogen om studies uit verschillende plaatsen en tijden te vergelijken, ongebruikelijke of opkomende viruslinies vroeg te signaleren en genetische patronen te koppelen aan reële ziektetrends. Op de lange termijn versterken tools als rhinotypeR het routinematig volgen van schijnbaar gewone verkoudheden die bij veel mensen ernstige ziekte kunnen uitlokken.
Bronvermelding: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Trefwoorden: rhinovirus-genotypering, moleculair toezicht, VP4/2-sequencing, bioinformatica-tools, luchtwegvirussen