Clear Sky Science · nl
Vergelijkende evaluatie van HTG- en TempO-Seq-gerichte transcriptoomprofileringmethoden
Waarom dit belangrijk is voor kankerzorg
Wanneer artsen en onderzoekers kanker bestuderen, richten ze zich vaak op de «boodschapper»-moleculen van de cel — RNA — om te zien welke genen actief of stil zijn. Deze patronen kunnen onthullen hoe een tumor zich gedraagt en welke behandelingen het beste kunnen werken. De meeste ziekenhuismonsters worden echter na formalinebehandeling in paraffine ingebed (FFPE) bewaard, wat het kwetsbare RNA beschadigt. Deze studie stelt een praktische vraag met grote gevolgen voor kankeronderzoek: nu een veelgebruikte RNA-test van de markt is verdwenen, kan een nieuwere methode deze vervangen en even nuttige resultaten leveren uit deze routinematig bewaarde monsters?
Twee instrumenten om genactiviteit te lezen
Jarenlang vertrouwden veel laboratoria op een methode genaamd HTG EdgeSeq Human Transcriptome Panel (HTP) om genactiviteit rechtstreeks te meten uit kleine schraapsels van FFPE-weefsel. Deze aanpak kon bijna alle menselijke genen onderzoeken zonder eerst RNA te extraheren, wat tijd bespaarde en kostbaar materiaal spaarde. Het bedrijf achter HTG EdgeSeq ging echter failliet, waardoor onderzoekers op zoek gingen naar een alternatief. Een nieuwere technologie, TempO-Seq (TOS) van een andere fabrikant, belooft vergelijkbare mogelijkheden: ook deze richt zich op veel genen tegelijk, werkt met beschadigd RNA uit FFPE-monsters en is ontworpen om gevoelig, reproduceerbaar en relatief betaalbaar te zijn.
De methoden aan de tand voelen
Het onderzoeksteam vergeleek deze twee technologieën rechtstreeks in een zeer praktische setting. Ze analyseerden 21 bewaarde endometriumkankermonsters, samen met drie standaard RNA-referentiematerialen, eerst met HTG HTP en daarna met TempO-Seq. Beide methoden gebruikten gerichte panelen die samen meer dan 18.000 van dezelfde genen besloegen. De wetenschappers voerden strikte kwaliteitscontroles uit om te waarborgen dat elk monster voldoende sequentielezingen opleverde en dat de metingen stabiel waren. Ze gebruikten ook statistische hulpmiddelen om "batch-effecten" te verwijderen — artificiële verschillen die simpelweg kunnen ontstaan doordat tests op verschillende dagen, machines of platforms zijn uitgevoerd.
Wat overeenkomt en wat niet
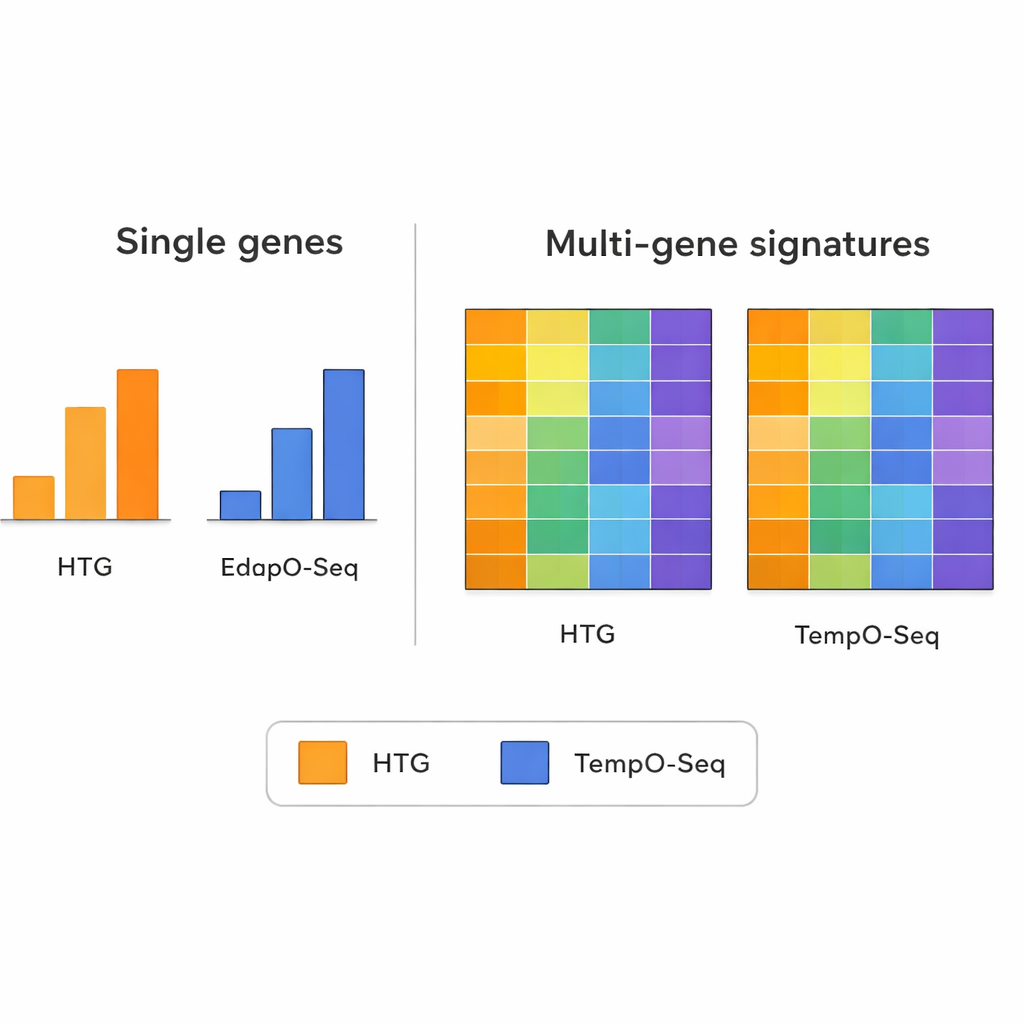
Toen het team keek naar de expressie van individuele genen één voor één, kwamen de twee methoden niet altijd overeen. Verschillen in hoe elke technologie zijn probes ontwerpt, monsters voorbereidt en lezingen telt, kunnen vergelijkingen op gen-niveau rumoerig maken. Dit beeld veranderde echter toen ze bredere patronen onderzochten die informatie van veel genen tegelijk combineren. Multi-genhandtekeningen — zoals die worden gebruikt om tumoren in moleculaire subtypes te groeperen, te schatten hoeveel immuuncellen in een monster aanwezig zijn, of in te schatten hoe zuiver het tumormateriaal is — lieten veel sterkere overeenstemming zien tussen TempO-Seq en HTG. In de meeste gevallen waren de scores of classificaties vergelijkbaar, zelfs nadat de onderzoekers simuleerden met minder sequentielezingen om verschillende machinemogelijkheden na te bootsen.
Multi-genpatronen als betrouwbare signalen
De studie benadrukt een belangrijk principe in de moderne genomica: terwijl de meting van een enkel gen door technische eigenaardigheden verstoord kan worden, heeft het combineren van signalen van tientallen of honderden genen de neiging die ruis uit te middelen. De auteurs gebruikten meerdere algemeen bekende multi-genhulpmiddelen als technische stresstests. Hieronder vielen een borstkankerpaneel dat tumoren toewijst aan intrinsieke subtypes, een algoritme dat scoort hoeveel immuun- en bindweefsel in een tumormonster is gemengd, en een methode die de verhoudingen van vele immuunceltypen schat. Over deze complexe uitkomsten volgde TempO-Seq over het algemeen nauwkeurig HTG, wat suggereert dat het dezelfde biologische verhalen opvangt, ook al verschillen sommige fijne details.
Wat dit betekent voor de toekomst
Voor onderzoekers die afhankelijk zijn van FFPE-archieven om kanker te bestuderen, had het verlies van een vertrouwd platform een grote tegenslag kunnen zijn. Deze benchmarkstudie biedt geruststelling: TempO-Seq lijkt een degelijke vervanger voor HTG HTP wanneer het doel is om multi-genbiomerkers en brede expressiepatronen te gebruiken, die de ruggengraat vormen van veel moderne diagnostische en prognostische hulpmiddelen. De auteurs waarschuwen dat het direct vergelijken van resultaten per enkel gen tussen platforms onverstandig is, omdat elke methode genen op iets verschillende manieren target. In plaats daarvan raden ze aan te focussen op complexe multi-genhandtekeningen voor cross-platformwerk. Simpel gezegd: de nieuwe methode lijkt het werk van zijn voorganger voor de meeste toepassingen in de oncologie te kunnen voortzetten, vooral wanneer onderzoekers geven om het algemene patroon van veel genen in plaats van de exacte waarde van slechts één gen.
Bronvermelding: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Trefwoorden: transcriptomische profilering, endometriumcarcinoom, FFPE-weefsel, gerichte RNA-sequencing, genexpressiebiomerkers