Clear Sky Science · nl
Een machine learning-benadering voor het voorspellen van osmotische coëfficiënten en het afleiden van activiteitcoëfficiënten in alkylammoniumzouten
Alledaagse chemicaliën met verborgen complexiteit
Van wasverzachters en haarconditioners tot desinfectiedoekjes en mondwater: een familie chemicaliën genaamd quaternaire ammoniumzouten — vaak verkort tot “Quats” — speelt op de achtergrond een belangrijke rol in veel producten waarop we vertrouwen. Ze helpen bij het doden van microben, het verzachten van textiel en het versnellen van industriële reacties. Toch is het verrassend moeilijk gebleken om precies te voorspellen hoe deze zouten zich in water gedragen, wat de efficiëntie van het ontwerpen van veiligere en groener formules beperkt. Deze studie laat zien hoe moderne machine learning kan leren van eerdere metingen om dat gedrag flexibeler en in veel gevallen nauwkeuriger te voorspellen dan traditionele modellen.
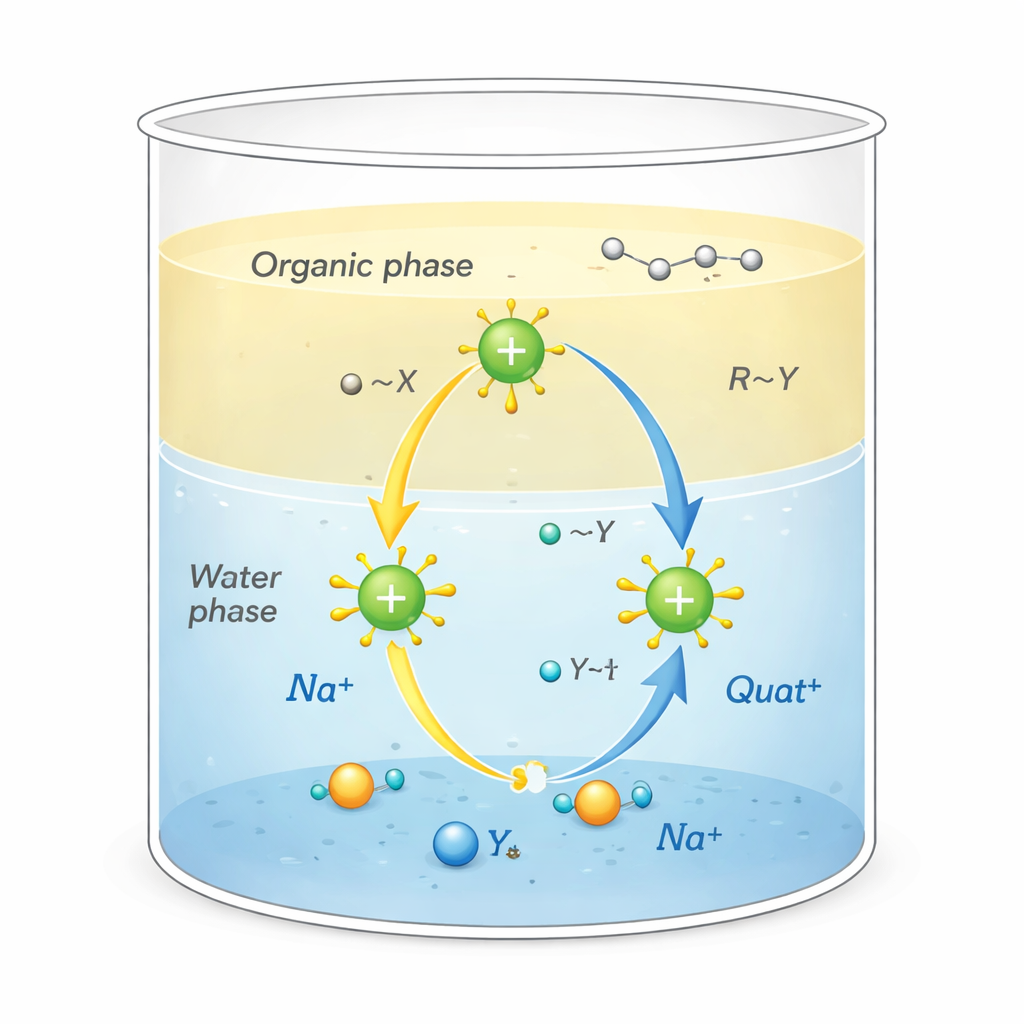
Waarom deze zouten van belang zijn
Quats zijn positief geladen moleculen omgeven door koolstofrijke “staarten”. Deze ongewone vorm stelt hen in staat meerdere taken tegelijk uit te voeren: zich hechten aan vettig vuil, blijven kleven aan oppervlakken zoals textiel of haar, en de membranen van microben verstoren, waardoor ze krachtige desinfectiemiddelen en oppervlakte-actieve stoffen zijn. Ze worden ook gebruikt als fase-overdrachtskatalysatoren, die fungeren als pendels die reactieve ionen van water naar olieachtige oplosmiddelen vervoeren waar ze normaal niet heen zouden gaan. Die pendelbeweging, die plaatsvindt op de grens tussen water en olie, kan chemische reacties die worden gebruikt bij de productie van farmaca, polymeren en fijnchemicaliën aanzienlijk versnellen.
Waarom het moeilijk is hun gedrag te voorspellen
Om nieuwe Quats te ontwerpen of bestaande te optimaliseren, moeten chemici weten hoe ze zich in oplossing gedragen — hoe sterk ze met water en met andere opgeloste ionen interageren. Twee belangrijke grootheden zijn de osmotische coëfficiënt, die weergeeft hoe zouten de neiging van water om door membranen te trekken beïnvloeden, en de activiteitscoëfficiënt, die aangeeft hoe “effectief” een opgeloste soort is vergeleken met een ideale, perfect gemengde oplossing. Traditioneel worden deze waarden verkregen door arbeidsintensieve experimenten of door het gebruik van complexe fysische modellen zoals Electrolyte-NRTL en Extended UNIQUAC, die veel afgestemde parameters vereisen en niet makkelijk te generaliseren zijn naar nieuwe moleculen.
Een computer leren moleculen te lezen
De onderzoekers kozen een andere route: ze vroegen zich af of een computer de relatie tussen Quat-structuur en osmotisch gedrag rechtstreeks uit bestaande data kon leren. Ze verzamelden 1.654 metingen van osmotische coëfficiënten voor 52 verschillende Quats uit de wetenschappelijke literatuur. Elk molecuul werd beschreven met SMILES-notatie — een stringrepresentatie die kenmerken codeert zoals het aantal koolstof- en zuurstofatomen, de aanwezigheid van benzeenringen, vertakkingen en het type positief geladen stikstofgroep, samen met het bijbehorende negatieve ion (zoals chloride, bromide of nitraat). Deze structurele descriptoren, plus de zoutconcentratie, dienden als invoer voor meerdere supervised machine-learningalgoritmen die in Python werden geïmplementeerd.
Het meest betrouwbare voorspellende model vinden
Zeven verschillende algoritmen — waaronder lineaire regressie, beslissingsbomen, random forests, support vector machines, gradient boosting, k-nearest neighbors en Gaussian processes — werden getraind op 70% van de data en getest op de resterende 30%. Het team gebruikte ook een striktere validatiestrategie waarbij alle data voor één zout werden weggelaten om te zien hoe goed de modellen extrapoleerden naar een daadwerkelijk onzichtbare verbinding. Lineaire regressie presteerde slecht en miste belangrijke niet-lineaire trends. Boomgebaseerde methoden pasten zich extreem goed aan de trainingsdata aan maar leverden licht gekartelde voorspellingen en verloren nauwkeurigheid bij nieuwe zouten. Het Gaussian process-model vond de beste balans: het leverde soepele, fysisch redelijke krommen voor osmotische coëfficiënten en behaalde een gemiddelde absolute procentuele fout van ongeveer 5% in totaal, waarmee het andere machine-learningbenaderingen overtrof bij de zwaarste tests.
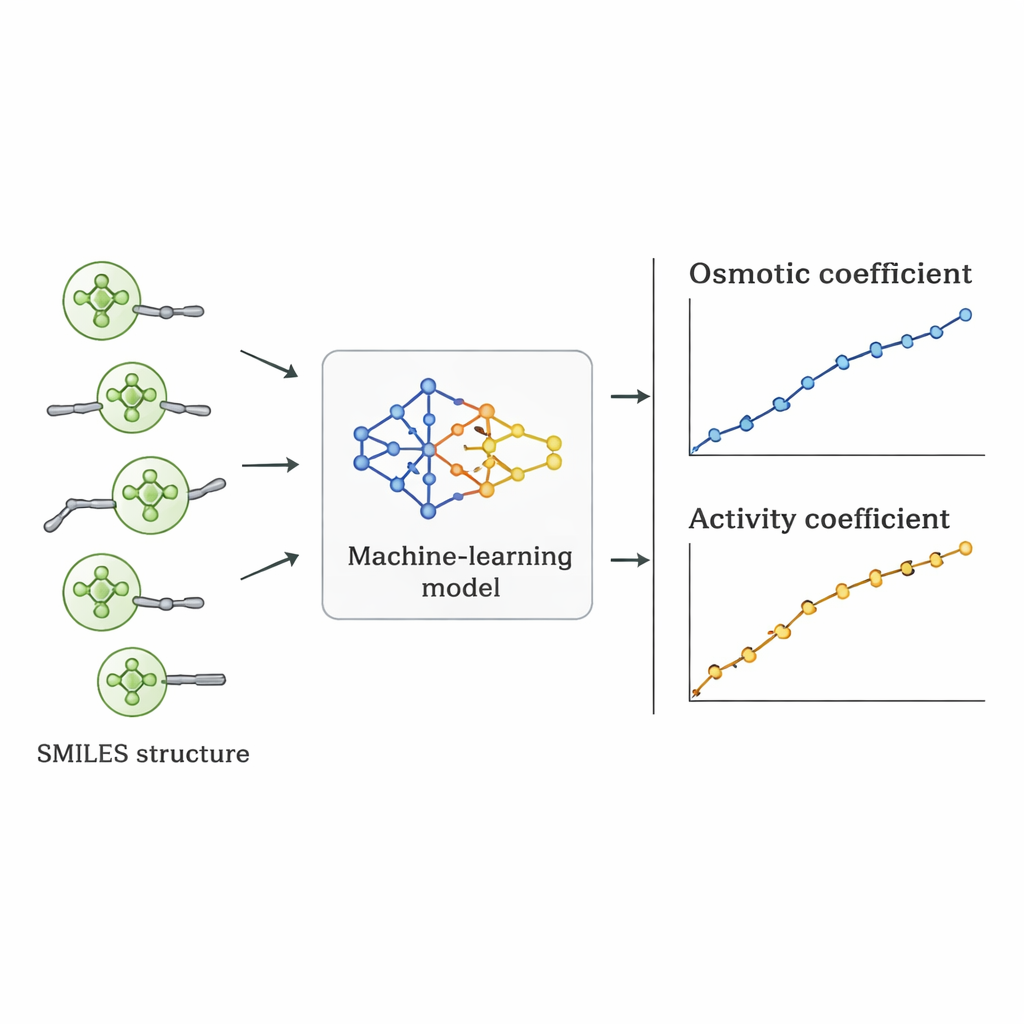
Van osmotisch gedrag naar bruikbare ontwerpcijfers
Zodra het beste model was gekozen, werden de voorspelde osmotische coëfficiënten omgezet in activiteitscoëfficiënten met gebruik van standaard thermodynamische relaties. Wanneer deze activiteitscoëfficiënten werden vergeleken met waarden afgeleid uit experimenten en uit gevestigde fysische modellen, evenaarde of overtrof de machine-learningbenadering die waarden vaak voor individuele Quats. Hoewel de gemiddelde fout over alle stoffen iets hoger was dan bij sommige gespecialiseerde modellen, had het een cruciaal voordeel: omdat het wordt aangestuurd door structurele descriptoren in plaats van zout-specifieke afstemming, kan het worden toegepast op nieuwe Quats die nooit in het laboratorium zijn gemeten, zolang hun structuren lijken op die in de trainingsset.
Wat dit betekent voor producten en processen
Voor een niet-specialist is de boodschap dat computers nu compacte tekstbeschrijvingen van moleculen kunnen “lezen” en op basis van patronen geleerd uit eerdere data kunnen voorspellen hoe die moleculen zich in water zullen gedragen met indrukwekkende nauwkeurigheid. Dit opent de deur naar snellere, goedkopere screening van nieuwe Quats voor desinfectiemiddelen, reinigingsmiddelen, persoonlijke verzorgingsproducten en industriële katalysatoren, zonder voor elke kandidaat uitputtend te hoeven experimenteren. Het huidige model is slechts een eerste stap, en de auteurs merken op dat rijkere moleculaire fingerprints en nieuwere algoritmen de prestaties verder kunnen verbeteren. Toch toont het aan hoe datagedreven hulpmiddelen traditionele chemie kunnen aanvullen, en ingenieurs kunnen helpen bij het ontwerpen van effectievere en mogelijk veiligere formules door chemische mogelijkheden te verkennen die onpraktisch zouden zijn om één voor één in het laboratorium te testen.
Bronvermelding: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Trefwoorden: quaternaire ammoniumzouten, fase-overdracht katalyse, osmotische coëfficiënten, activiteitscoëfficiënten, machine learning in chemie