Clear Sky Science · nl
Een kwantitatieve studie van cytotoxische verbindingen met grafgebaseerde descriptoren en machine learning
Waarom dit onderzoek van belang is voor toekomstige kankerbehandelingen
Kankergeneesmiddelen die tumorcellen doden, bekend als cytotoxische middelen, balanceren vaak tussen levensreddende effecten en ernstige bijwerkingen. Om veiligere en effectievere behandelingen te ontwerpen, hebben wetenschappers snelle en betrouwbare methoden nodig om te voorspellen hoe deze middelen zich door het lichaam bewegen—hoe goed ze worden opgenomen, hoe gemakkelijk ze celmembranen passeren en waar ze terechtkomen. Deze studie toont aan hoe wiskundige beschrijvingen van medicijnmoleculen, gecombineerd met moderne machine learning, nauwkeurig een sleutelkenmerk kunnen inschatten dat dit gedrag beheerst, wat het zoeken naar betere kankertherapieën kan versnellen.
Een cruciaal oppervlak dat bepaalt waar geneesmiddelen naartoe kunnen gaan
Een centraal idee in het artikel is de topologische polaire oppervlaktes (Top_PSA). In eenvoudige bewoordingen is dit een getal dat aangeeft hoeveel van het oppervlak van een molecule uit "polaire" gebieden bestaat—delen die van water houden en waterstofbruggen kunnen vormen. Moleculen met zeer grote polaire oppervlakten hebben vaak moeite met het passeren van vetachtige celmembranen en kunnen slecht worden opgenomen bij orale inname. Moleculen met zeer kleine polaire oppervlakten kunnen juist te gemakkelijk door barrières glippen, waardoor ze soms ongewenste bijwerkingen in gevoelige weefsels zoals de hersenen veroorzaken. Top_PSA is een populair snelkader voor het schatten van deze transporteigenschappen omdat het snel berekend kan worden uit een 2D-tekening van een molecule, zonder langzame 3D-simulaties.
Moleculaire tekeningen omzetten in cijfers
De onderzoekers stelden een zorgvuldig samengestelde set van 156 verschillende cytotoxische verbindingen samen, afkomstig uit echte anticancerogene middelen en experimentele stoffen. Ze zetten vervolgens elk molecuul om in 58 zogeheten descriptoren—getallen die eigenschappen vastleggen zoals het aantal atomen, het aantal ringen, hoe flexibel de bindingen zijn, hoeveel atomen waterstofbruggen kunnen vormen, en hoe polair of elektronegatief verschillende delen zijn. Veel van deze descriptoren komen uit de grafentheorie, waarbij een molecule wordt behandeld als een netwerk van verbonden knooppunten en verbindingen. Dit rijke numerieke portret van elk molecuul diende als invoer voor computermodellen die de Top_PSA-waardes proberen te voorspellen zoals berekend door veelgebruikte chemie-toolkits.
Meerdere wegen naar nauwkeurige voorspelling testen
Om de beste manier te vinden om deze descriptoren aan Top_PSA te koppelen, vergeleek het team verschillende modelleringsstrategieën. Ze probeerden standaard lineaire regressie en twee "geregulariseerde" versies genaamd ridge- en LASSO-regressie, die beter omgaan met ruis en overlappende informatie. Ze onderzochten ook verschillende methoden voor datavoorbewerking: modellen direct op de ruwe descriptoren passen, deze comprimeren met principale componentenanalyse (PCA), ze schalen op een manier die de invloed van extreme waarden vermindert (robust scaling), uitbijters aanpassen en sterk gecorreleerde kenmerken verwijderen met een maat genaamd de variance inflation factor. Elke aanpak werd zorgvuldig beoordeeld met k-fold cross-validatie, een methode die de data herhaaldelijk in trainings- en testsubsets splitst om overfitting te voorkomen.
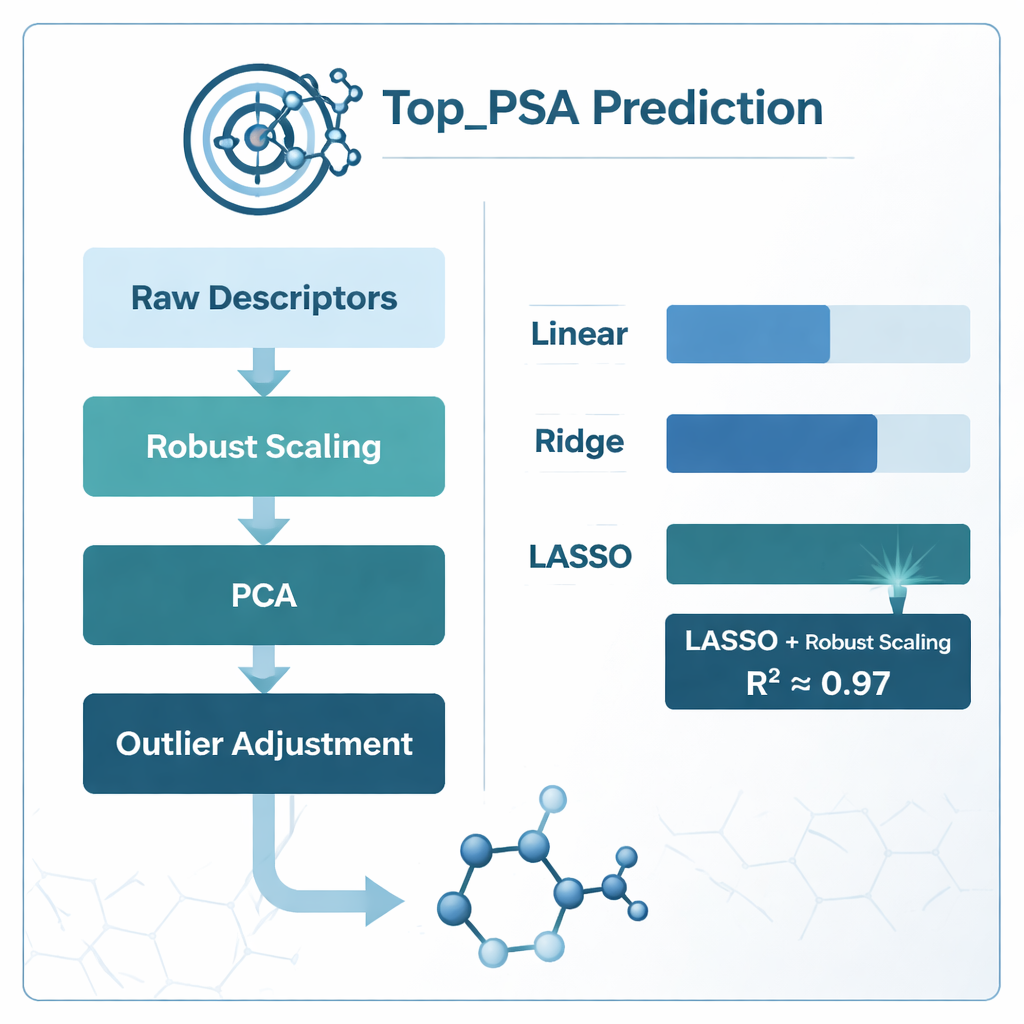
Wat het beste werkte en wat de modellen leerden
De duidelijke winnaar was de combinatie van robust scaling met LASSO-regressie, die een determinatiecoëfficiënt (R²) van ongeveer 0,97 behaalde—wat betekent dat het model ruwweg 97% van de variatie in Top_PSA over de 156 middelen kon verklaren. PCA-gebaseerde modellen kwamen qua ruwe nauwkeurigheid dichtbij maar waren chemisch moeilijker te interpreteren omdat de oorspronkelijke descriptoren vermengd raken tot abstracte componenten. Eenvoudig verwijderen van gecorreleerde descriptoren met de variance inflation factor schaadde de prestaties zelfs, wat suggereert dat enige overlap nog steeds nuttige chemische informatie bevat. Door te kijken welke descriptorgewichten LASSO niet op nul zette, vonden de auteurs dat de belangrijkste factoren de aanwezigheid van heteroatomen zoals stikstof en zuurstof waren, de mogelijkheid om waterstofbruggen te geven of te accepteren, en indices die bijhouden hoe elektronegatieve atomen over de moleculaire graf zijn verdeeld—allemaal kenmerken die overeenkomen met het intuïtieve chemische begrip van polair oppervlak.
Hoe dit beter geneesmiddeldesign kan sturen
Voor lezers buiten het vakgebied is de kernboodschap dat zorgvuldig voorbereide wiskundige vingerafdrukken van moleculen, gecombineerd met goed gekozen machine-learningmethoden, snelle en betrouwbare schattingen kunnen geven van hoe "plakkend" of "glad" kankergeneesmiddelen zich door het lichaam bewegen. De studie biedt praktische richtlijnen voor andere onderzoekers over hoe descriptorgegevens voor te verwerken, welke modelleringsbenaderingen te verkiezen en welke snelkoppelingen te vermijden. Op de lange termijn kunnen zulke robuuste, interpreteerbare modellen van Top_PSA chemici helpen enorme virtuele bibliotheken met potentiële geneesmiddelen te filteren, zodat ze zich concentreren op verbindingen met de juiste balans tussen membraanpassage en veiligheid—een belangrijke stap richting effectievere en minder toxische kankerbehandelingen.
Bronvermelding: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Trefwoorden: cytotoxische geneesmiddelen, polair oppervlak, moleculaire descriptoren, machine learning, geneesmiddelpermeabiliteit