Clear Sky Science · nl
Mitochondriale disfunctie en Ca2+-dysregulatie in menselijk iPSC-afgeleide neuronen met preseniline-1-mutatie ontstaan onder stress via een MCU-1-onafhankelijk mechanisme
Waarom dit belangrijk is voor de ziekte van Alzheimer
De ziekte van Alzheimer wordt vaak beschreven aan de hand van plakkerige eiwitplaques in de hersenen, maar lang voordat het geheugen achteruitgaat, kunnen de kleine "energiecentrales" in zenuwcellen — de mitochondriën — en de afhandeling van calciumionen al fout gaan. Deze studie gebruikt menselijke neuronen gekweekt uit huidcellen van een persoon die drager is van een bekende familiale Alzheimer-mutatie om een eenvoudige maar cruciale vraag te stellen: hoe vroeg, en op welke manier, beginnen energieproductie en calciumbalans te falen?
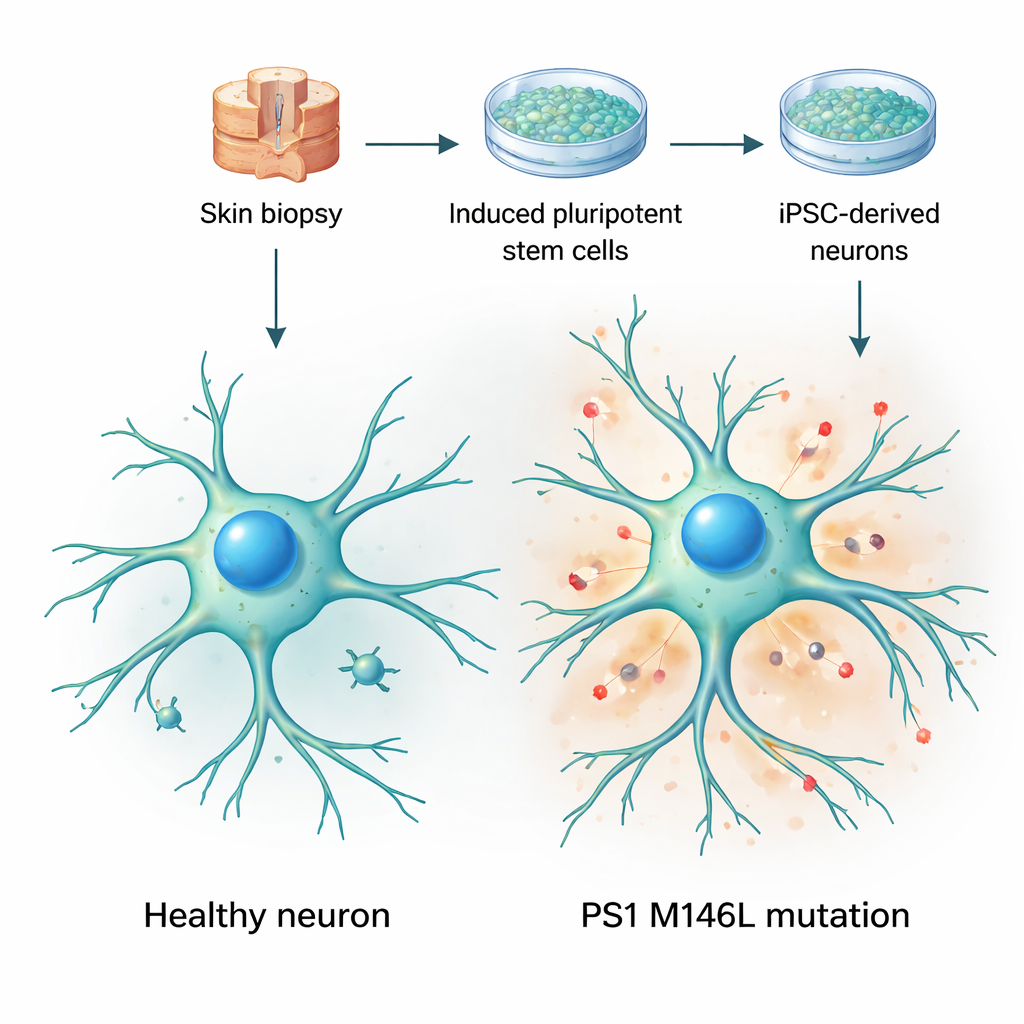
Huidcellen omzetten in levende breinmodellen
De onderzoekers begonnen met huidbiopten van twee vrouwen: een gezonde vrijwilligster en een symptoomvrije drager van een preseniline-1-mutatie genaamd M146L, die voorkomt in een Argentijnse familie met vroeg voorkomende Alzheimer. Ze programmeerden de huidcellen terug naar geïnduceerde pluripotente stamcellen — cellen die bijna elk weefsel kunnen worden — en leidden ze vervolgens naar zenuwcellen. Gedurende enkele weken in kweek kregen deze cellen kenmerkende neuronale vormen, verlengden lange vertakte uitlopers en brachten gebruikelijke neuronale markers tot expressie. Belangrijk is dat zowel de controle- als de mutantecellen in gelijke mate rijpten en er over het algemeen gezond uitzagen, wat het team in staat stelde zich te richten op subtiele functionele veranderingen in plaats van op duidelijk celverlies of schade.
Elektrische signalen en calcium onder druk
Zenuwcellen zijn afhankelijk van strikte controle van calcium, een geladen atoom dat fungeert als een snelle aan/uit-schakelaar voor veel cellulaire processen. Met behulp van fluorescerende kleurstoffen volgde het team hoe calciumniveaus in de cellen veranderden wanneer ze elektrisch werden gestimuleerd met kalium of geactiveerd met signaalmoleculen. Onder eenvoudige depolariserende stimulatie lieten neuronen met de M146L-mutatie zwakkere calciumstijgingen zien dan controleneuronen, wat wijst op problemen met het behouden van de elektrische en ionische gradiënten die normaal calciumintocht aandrijven. Toen de onderzoekers echter een meer stressvolle situatie opwekten — door calcium uit interne opslagplaatsen in het endoplasmatisch reticulum te laten lekken — werd het verschil duidelijker. Als reactie op deze stress namen de mitochondriën in mutante neuronen merkbaar minder calcium op dan die in controlecellen, wat wijst op een verminderde capaciteit om gevaarlijke calciumpieken te bufferen.
Ontkoppeling van energieverbruik en calciumbalans
Om te begrijpen hoe deze veranderde calciumregulatie de celstofwisseling beïnvloedt, maten de onderzoekers hoeveel zuurstof de neuronen verbruikten — een directe proxy voor mitochondriale activiteit. Verrassend genoeg ademden neuronen met de M146L-mutatie zwaarder: hun basale en maximale zuurstofconsumpties en de hoeveelheid zuurstof gekoppeld aan ATP-productie waren allemaal hoger dan bij controlecellen. Toch leek de efficiëntie waarmee zuurstofgebruik aan ATP-productie werd gekoppeld vergelijkbaar, en er was geen toename in het aantal mitochondriën of in sleutelenzymen voor ATP-productie. In plaats daarvan waren mitochondriën in mutante neuronen langer en meer buisvormig, met hogere niveaus van een fusie-eiwit genaamd mitofusine-1, een patroon dat vaak wordt gezien bij cellen onder chronische, laaggradige stress. Deze hyperactieve, verlengde mitochondriën genereerden ook meer reactieve zuurstofsoorten, onstabiele moleculen die eiwitten en DNA kunnen beschadigen als ze niet goed worden gecontroleerd.
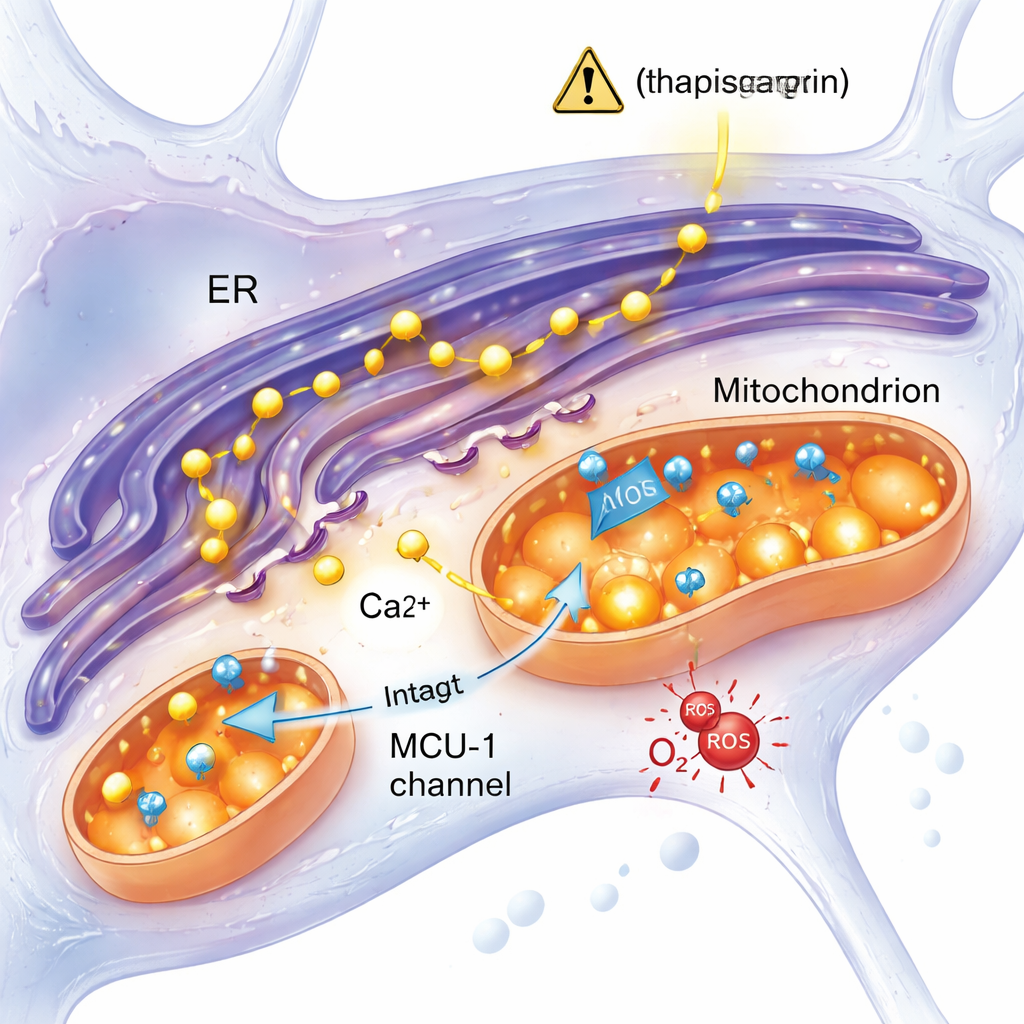
Een stressreactie onafhankelijk van een belangrijke calciumkanaal
Een toonaangevende hypothese in Alzheimer-onderzoek veronderstelt dat overtollig calcium uit het endoplasmatisch reticulum via een kanaal genaamd de mitochondriale calciumuniporter (MCU-1) de mitochondriën binnenstroomt, ze overbelast en disfunctie veroorzaakt. Deze studie testte die veronderstelling rechtstreeks. Toen het team MCU-1 blokkeerde met een specifieke remmer, lieten zowel controle- als mutante neuronen sterke verminderingen in mitochondriale calciumopname zien, wat bevestigt dat het kanaal zelf in beide groepen functioneerde. Bovendien, wanneer calciumafgifte werd opgewekt via een meer fysiologische route waarbij de IP3-receptor betrokken is — een andere sleutelpoort voor calcium — reageerden de mutante en controlecellen vergelijkbaar. Deze resultaten wijzen weg van een defect MCU-1-kanaal en suggereren in plaats daarvan dat de fysieke en functionele contacten tussen het endoplasmatisch reticulum en de mitochondriën, of andere aspecten van hun interactie, veranderd zijn in de mutante neuronen.
Wat dit betekent voor begrip en behandeling van de ziekte
Samengevoegd schetsen de bevindingen het beeld van menselijke neuronen met de PS1 M146L-Alzheimer-mutatie als cellen die in rust normaal lijken maar abnormaal reageren onder stress. Hun mitochondriën nemen niet genoeg calcium op wanneer interne voorraden plotseling vrijkomen, en toch lopen ze heter — ze verbruiken meer zuurstof en produceren meer reactieve zuurstofsoorten — alsof ze vastzitten in een kostbare compenserende modus. Omdat dit gebeurt in levende, menselijk-afgeleide neuronen vóór klinische symptomen, ondersteunt het werk het idee dat verstoorde calciumsignalering en vroege mitochondriale overbelasting upstream-gebeurtenissen zijn in Alzheimer, en niet alleen late bijproducten. Voor niet-specialisten is de kernboodschap dat het bewaren van de balans tussen calciumsignalen en mitochondriale energieproductie wellicht even essentieel kan zijn voor het voorkomen van de ziekte als het richten op de beter bekende amyloïde plaques.
Bronvermelding: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Trefwoorden: Ziekte van Alzheimer, mitochondriën, calciumsignalering, preseniline-1-mutatie, iPSC-afgeleide neuronen