Clear Sky Science · nl
Stochastische LASSO voor extreem hoog-dimensionale genomische data
De naalden vinden in genomische hooibergen
De moderne biologie kan tienduizenden genen tegelijk meten, maar patiëntonderzoeken bevatten vaak slechts enkele honderden mensen. In deze scheefheid zitten kleine groepen genen die echt van belang zijn voor het voorspellen van ziekte‑risico of overleving. Dit artikel introduceert “Stochastische LASSO”, een statistische methode die is ontworpen om die sleutelgenen betrouwbaar te ontdekken in oceaanen van ruisende genomische data, zelfs wanneer er veel meer genen zijn dan patiënten.
Waarom het kiezen van de juiste genen zo moeilijk is
Onderzoekers vertrouwen vaak op instrumenten zoals LASSO, die onbelangrijke gen-effecten naar nul terugdringen terwijl ze de meest informatieve behouden. Klassieke versies van LASSO hebben echter moeite wanneer het aantal genen het aantal monsters ver overstijgt, zoals vaak voorkomt in kanker-genomica. Standaard LASSO kan hooguit evenveel genen selecteren als er patiënten zijn, en het neigt ernaar genen over het hoofd te zien die zich op vergelijkbare wijze gedragen. Eerdere verbeteringen die extra straffen toevoegen kunnen met enige correlatie omgaan, maar ze kunnen ook de biologische betekenis vervagen door gerelateerde genen te dwingen te handelen alsof ze allemaal in dezelfde richting op de uitkomst werken.
Het bouwen van schonere willekeurige monsters
Een veelbelovende oplossing is om LASSO herhaaldelijk aan te passen op veel kleinere, willekeurig geselecteerde subsets van genen en vervolgens de resultaten te combineren. Toch kampen deze “bootstrap”-benaderingen nog steeds met drie problemen: gecorreleerde genen kunnen elkaar opheffen, veel genen worden zelden of nooit bemonsterd, en zuivere willekeur maakt de uiteindelijke selectie instabiel. Stochastische LASSO pakt deze problemen frontaal aan met een nieuw bemonsteringsschema dat correlatie‑gebaseerde bootstrapping heet. In plaats van genen willekeurig te kiezen, geeft het doelbewust de voorkeur aan genen die minder gecorreleerd zijn met die al gekozen zijn, wat leidt tot kleinere sets genen die veel onafhankelijker zijn. Het zorgt er ook voor dat elk gen even vaak gebruikt wordt over de bootstrap‑runs, zodat geen enkel gen onterecht genegeerd wordt. 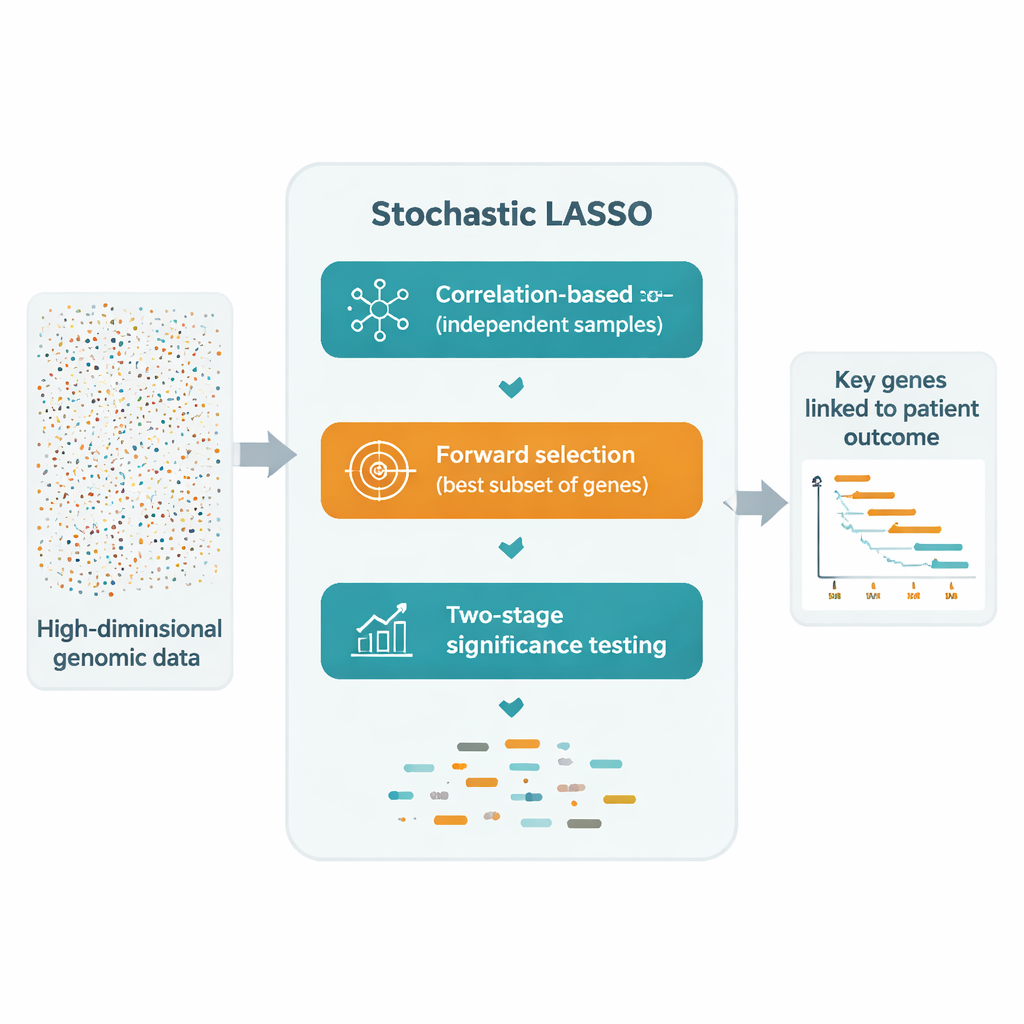
Van lokale aanwijzingen naar een globale genenset
Na het construeren van deze schonere subsets registreert Stochastische LASSO hoe groot de coëfficiënt van elk gen is over alle bootstrap‑fits. Dit gemiddelde van de absolute effecten wordt een “lokale score” die weerspiegelt hoe consistent belangrijk het gen blijkt te zijn. In plaats van elke mogelijke combinatie uitputtend te testen, bouwt de methode kandidaatmodellen door genen toe te voegen in volgorde van hun lokale scores en evalueert hoe goed elk kandidaatmodel uitkomsten voorspelt op aparte validatiegegevens. Op die manier komt het uit op een compacte set genen waarvan de gecombineerde signalen de data het beste verklaren, terwijl het veel minder proefnemingen vereist dan traditionele stapsgewijze methoden.
Testen welke genen echt van belang zijn
Om van “vaak geselecteerd” naar “statistisch overtuigend” te gaan, introduceren de auteurs een tweefasige t‑toets. Eerst controleren ze of de gemiddelde coëfficiënt van elk gen over de bootstraps duidelijk verschilt van nul, en markeren het als mogelijk betekenisvol. Vervolgens vragen ze onder deze kandidaten of het effect van elk gen groter is dan de typische effectgrootte van alle kandidaten. Alleen genen die beide toetsen doorstaan worden als significant verklaard. Omdat deze toetsen steunen op de vele bootstrap‑schattingen, kan Stochastische LASSO met vertrouwen meer significante genen identificeren dan er patiënten zijn — iets wat conventionele LASSO niet kan. 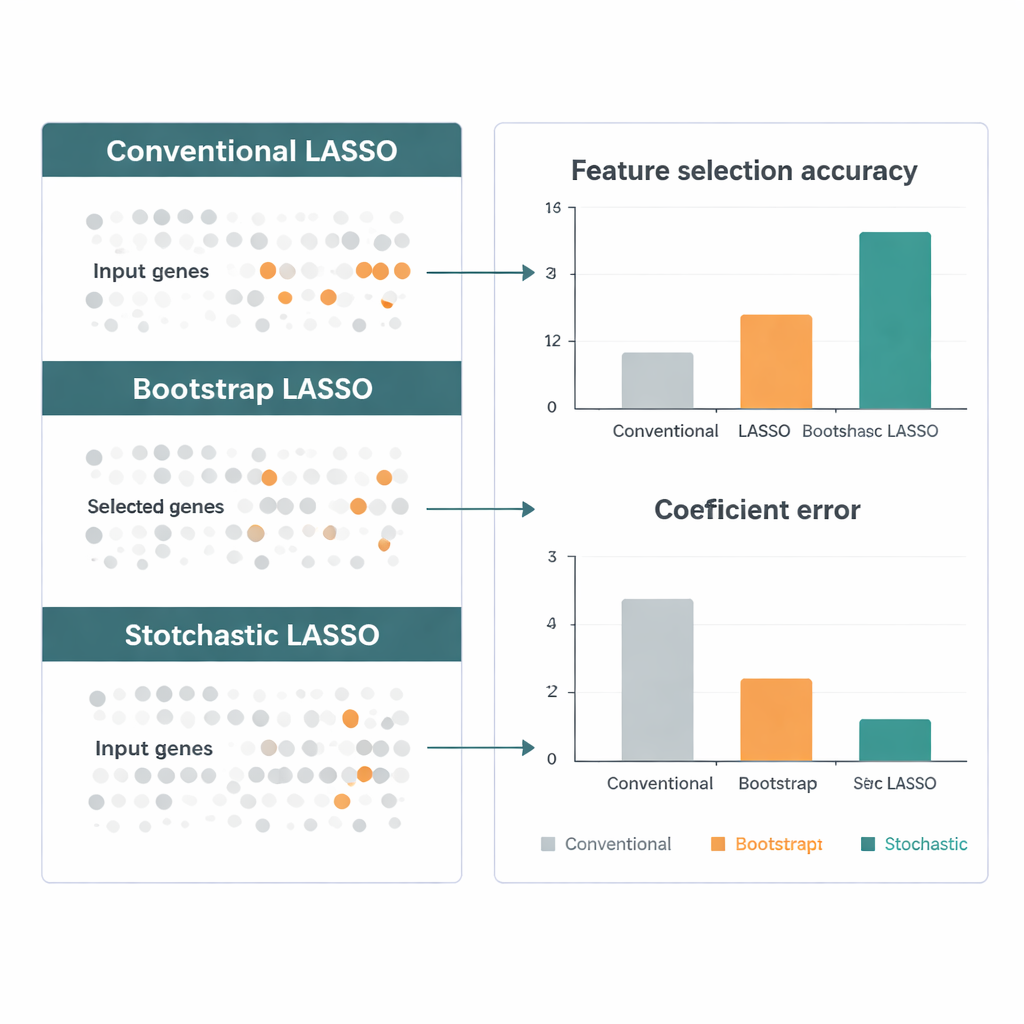
De waarde aantonen in simulaties en kankergegevens
De auteurs vergelijken Stochastische LASSO met verschillende toonaangevende LASSO‑varianten met behulp van gesimuleerde data die echte genomische studies nabootsen: zeer veel genen, sterke correlaties en bekende “echte” signalen. Over meerdere scenario’s vindt de nieuwe methode vaker de juiste genen, schat hun effecten nauwkeuriger en blijft stabiel van run tot run. Daarna richten ze zich op genexpressiegegevens uit The Cancer Genome Atlas voor hersentumoren, waaronder agressieve glioblastomen. Stochastische LASSO benadrukt honderden genen waarvan de activiteit samenhangt met patiëntoverleving en markeert biologische pathways — zoals signaalroutes en geneesmiddelmetabolisme — die onafhankelijke steun in de literatuur hebben, wat suggereert dat de methode niet alleen statistisch scherper is maar ook biologisch zinnig.
Wat dit betekent voor patiënten en onderzoekers
Voor niet‑specialisten is de kernboodschap dat Stochastische LASSO een slimmer filter is voor genomische big data. Het helpt wetenschappers echte ziektegerelateerde genen te scheiden van statistische ruis, zelfs wanneer data beperkt zijn en genen sterk met elkaar verbonden zijn. Door nauwkeurigere en stabielere genlijsten en effectschattingen te leveren, kan het de zoektocht naar biomarkers, geneesmiddeldoelen en prognostische signaturen bij kanker en andere complexe ziekten aanscherpen. Hoewel het is gedemonstreerd voor lineaire regressie, kan hetzelfde raamwerk worden ingebed in overlevingsmodellen en classificatieproblemen, waarmee het potentiële bereik binnen biomedisch onderzoek wordt vergroot.
Bronvermelding: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Trefwoorden: selectie van genomische kenmerken, hoog-dimensionale data, LASSO-methoden, kanker genexpressie, biomarkerontdekking