Clear Sky Science · nl
Minimale virtuele dataset voor reproduceerbare triploïde de novo-genoomassemblage
Waarom genomen met drie kopieën ertoe doen
Veel gewassen en andere organismen dragen niet slechts twee kopieën van elk chromosoom, zoals mensen dat doen — ze kunnen drie of meer kopieën hebben. Het reconstrueren van deze extra kopieën uit DNA-sequencinggegevens is verrassend lastig, omdat de kopieën zeer gelijkend maar niet volledig identiek zijn. Dit artikel introduceert een kleine maar zorgvuldig ontworpen “virtuele” dataset waarmee onderzoekers genoomassemblage-software kunnen testen en vergelijken op een realistisch drie-kopie (triploïde) probleem, onder volledig bekende en reproduceerbare condities.
Het bouwen van een eenvoudige plaatsvervangend genoom
In plaats van te beginnen met een echte plant of dier maakt de auteur eerst een willekeurige DNA-reeks van één miljoen letters als een schoon sjabloon. Dit sjabloon wordt vervolgens gedupliceerd in drie afzonderlijke versies, die de drie chromosoomsets in een triploïde organisme voorstellen. Om na te bootsen hoe echte genomen in de loop van de tijd langzaam veranderen, introduceert de studie een vast aantal kleine veranderingen — enkel-letter substituties — stap voor stap in elke kopie. Het herhalen van dit proces over 100 stappen produceert triplets van genomen die variëren van bijna identiek tot duidelijk maar nog altijd gematigd verschillend. Deze gecontroleerde “divergentiegradiënt” vormt de ruggengraat van de benchmark.
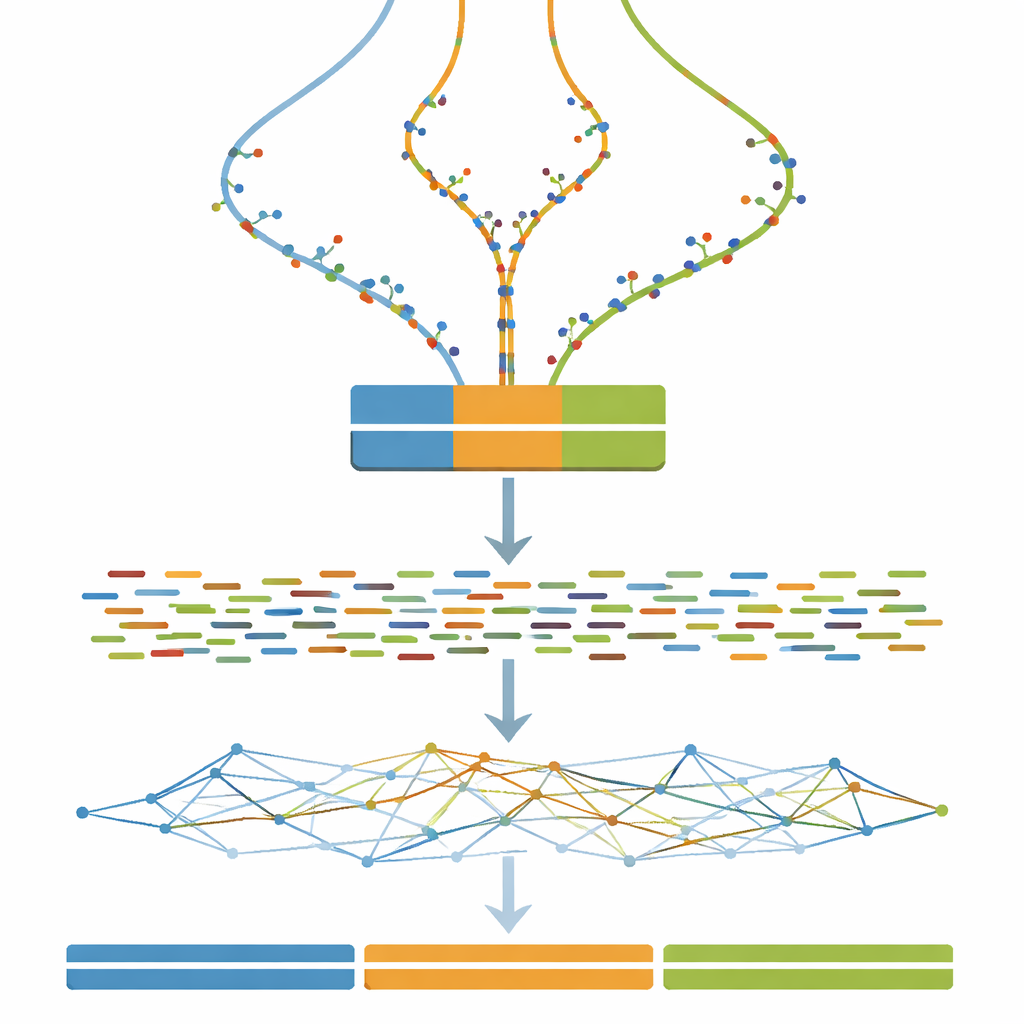
Virtuele genomen omzetten in virtuele experimenten
Zodra elk drie-kopie genoom gedefinieerd is, is de volgende stap het imiteren van wat een DNA-sequencer zou zien. De studie gebruikt veelgebruikte software om korte gepaarde DNA-fragmenten te simuleren, vergelijkbaar met die van een Illumina-sequencer, bij een constante en redelijk hoge dekking. Optionele opschoningsstappen bootsen gangbare praktijken in de echte wereld na, zoals het corrigeren van willekeurige sequencingfouten en het samenvoegen van overlappende read-paren. Daardoor kan iedereen die de dataset gebruikt niet alleen hun assemblage-algoritmen testen, maar ook zien hoe typische voorbewerkingskeuzes de uiteindelijke geassembleerde genomen beïnvloeden.
Assembly-strategieën op de proef stellen
De kern van het werk is een uitgebreid experiment waarbij alle gesimuleerde reads in één genoomassemblageprogramma worden gevoerd terwijl slechts één belangrijke instelling wordt veranderd: de k-mer grootte, een parameter die bepaalt hoe fijnmazig de software de reads ‘opdeelt’ bij het reconstrueren van het genoom. Voor elke combinatie van divergenceniveau (van 0 tot 100 stappen) en k-mer grootte (een breed scala aan oneven waarden) wordt een nieuwe assemblage gebouwd. Een begeleidend evaluatietool meet vervolgens hoe continu de verzamelde stukken zijn, hoeveel stukken er bestaan en hoe nauwkeurig hun gecombineerde lengte overeenkomt met de bekende drie miljoen letters tellende waarheid. Deze metingen worden samengevat in heatmaps, die brede zones onthullen waar assemblages verschillende kopieën samenvoegen, fragmenteren in veel kleine stukjes of dicht bij het ideaal van drie lange, nauwkeurige contigs komen.
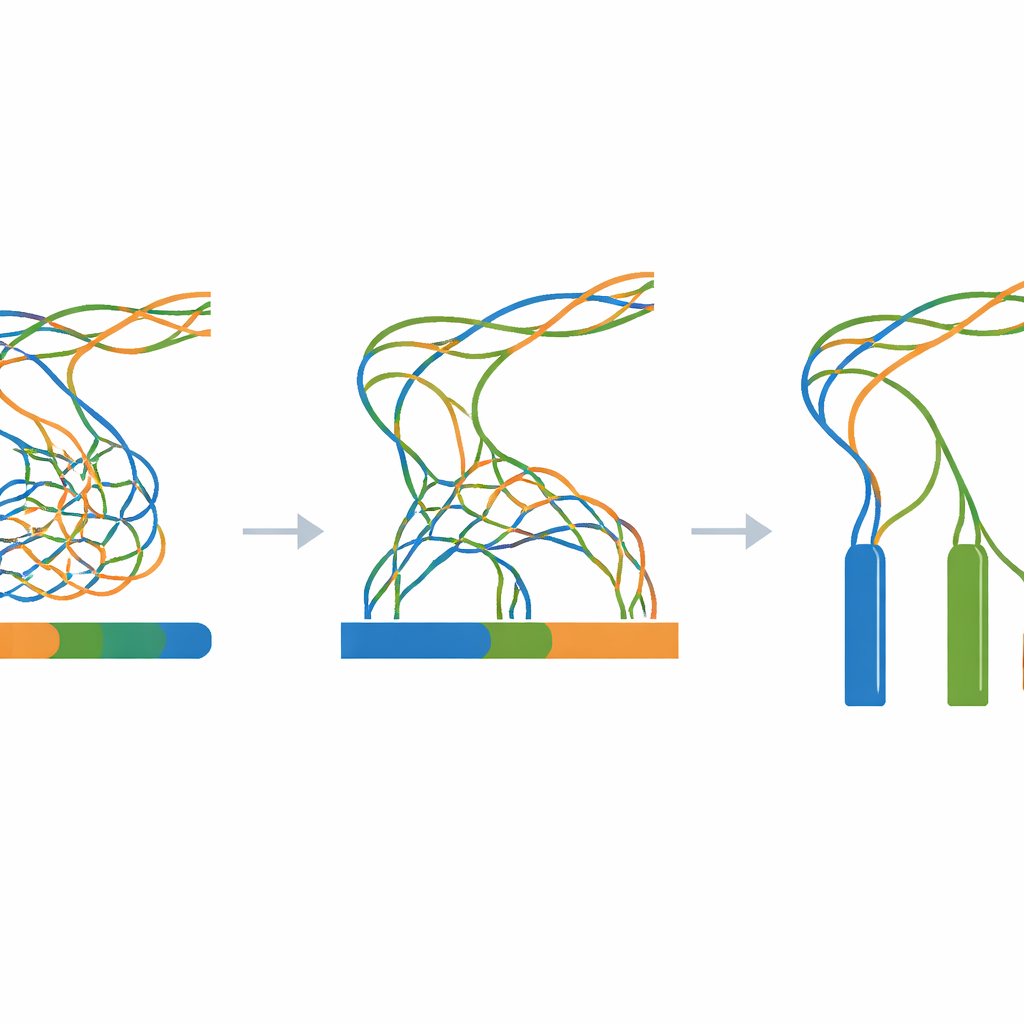
Een transparante referentie voor lastige genomen
Omdat elke stap synthetisch en gescript is — van het initiale willekeurige sjabloon tot de uiteindelijke assemblages — kunnen onderzoekers de volledige workflow reproduceren op een standaard Linux-computer met alleen open-source tools. Het Zenodo-archief dat in het artikel gelinkt is bevat het sjabloon-genoom, alle tussenliggende gemuteerde sequenties, alle gesimuleerde reads en elk assemblageresultaat, samen met logbestanden en eenvoudige hulpscripts. Technische controles bevestigen dat het mutatieproces zich gedraagt zoals verwacht, dat de gesimuleerde reads overeenkomen met de gevraagde lengtes en dekking, en dat assemblages het verwachte patroon tonen: sterke over-collapsing wanneer de drie kopieën bijna identiek zijn, en duidelijkere scheiding naarmate ze verder uit elkaar drijven.
Wat dit in gewone taal betekent
In gewone bewoordingen biedt dit artikel een gecontroleerde testbaan voor software die probeert drie vergelijkbare instructieboeken te herbouwen uit stapels door elkaar gehusselde fragmenten. Door geleidelijk te vergroten hoe verschillend de drie boeken zijn, en door systematisch een belangrijke instelling in het reconstructieproces te veranderen, maakt de dataset het eenvoudig te zien wanneer en hoe huidige methoden falen of slagen. Ontwikkelaars kunnen het gebruiken om nieuwe algoritmen te verfijnen, terwijl gebruikers beter begrijpen welke instellingen het beste werken voor triploïde genomen. Hoewel het DNA zelf kunstmatig is, zijn de lessen die het mogelijk maakt — over samenvoegen, scheiding en de impact van parameterkeuzes — direct relevant voor realistische inspanningen om de complexe genomen van veel belangrijke soorten te ontsleutelen.
Bronvermelding: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Trefwoorden: triploïde genoomassemblage, polyploïde benchmarking, synthetische DNA-dataset, de novo assemblage, k-mer optimalisatie