Clear Sky Science · nl
DECODE: op deep learning gebaseerde algemene deconvolutieframework voor diverse omics-gegevens
Waarom dit onderzoek ertoe doet
De moderne biomedische wetenschap zwemt in metingen van onze weefsels: welke genen actief zijn, welke eiwitten aanwezig zijn en welke kleine moleculen onze cellen van brandstof voorzien. Toch worden de meeste van deze metingen uitgevoerd op gemengde monsters, waarin veel celtypen door elkaar zitten. De studie achter DECODE introduceert een krachtig kunstmatig-intelligentieframework dat deze signalen kan ontmengen en ons kan vertellen welke cellen en celtoestanden aanwezig zijn, zelfs in zeer uiteenlopende soorten data. Deze mogelijkheid kan onderzoek naar kanker, immuniteit en metabole ziekten versnellen en tegelijk beter gebruik maken van bestaande biobankmonsters.
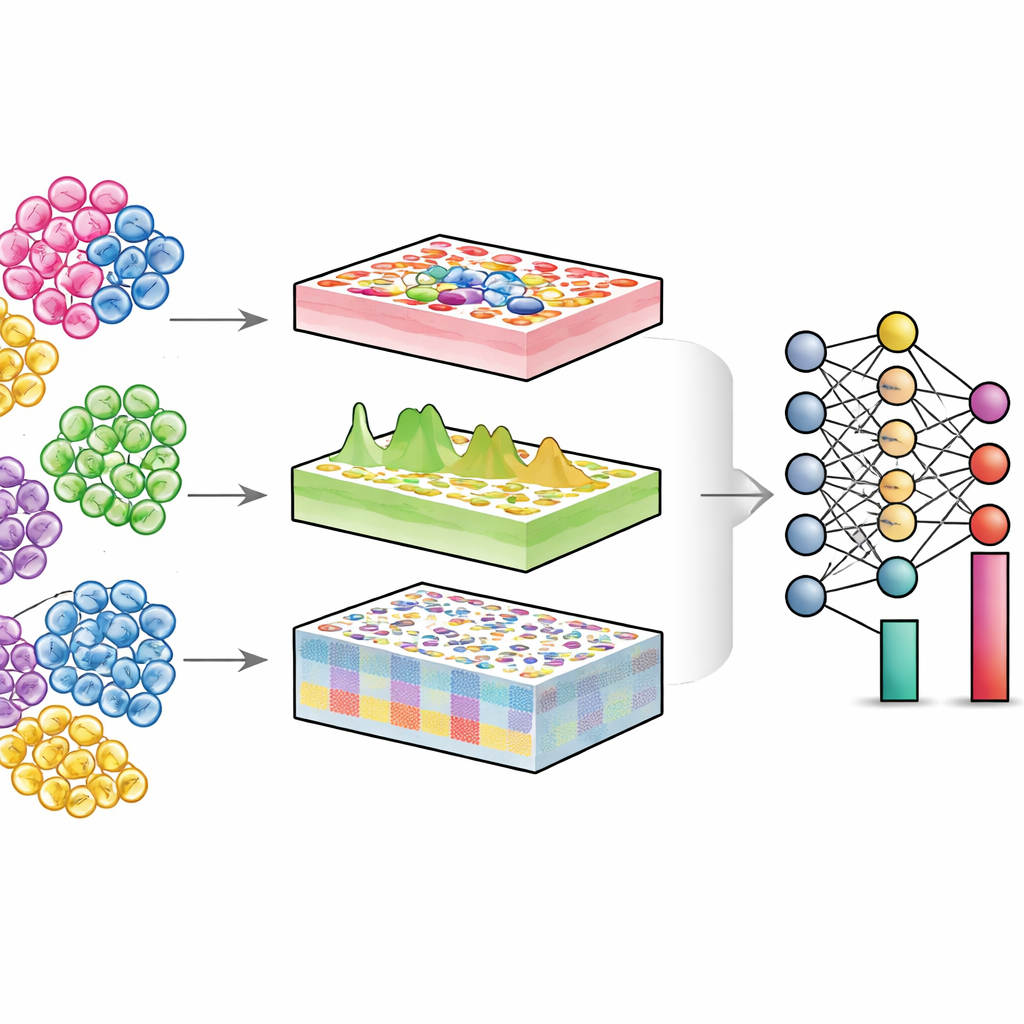
Inkijk in gemengde weefsels
Elk orgaan is een gemeenschap van verschillende celtypen—immuuncellen, structurele cellen, stamcellen en meer. In gezondheid en ziekte verandert vaak niet alleen wat elke cel doet, maar ook hoeveel van elk type aanwezig zijn en in welke toestand ze verkeren. Single-cell-technologieën kunnen individuele cellen direct meten, maar zijn duur en technisch veeleisend, vooral voor grote patiëntengroepen of oude opgeslagen monsters. Conventionele “bulk”-experimenten daarentegen mengen duizenden of miljoenen cellen en leveren een gemiddelde signaal. Deconvolutie-algoritmen proberen dit mengen ongedaan te maken: gegeven bulkdata en een referentiekaart van single cells, schatten ze de proportie van elk celtype in het weefsel.
De beperkingen van eendimensionale tools
Bestaande deconvolutiehulpmiddelen zijn meestal toegespitst op één soort meting, zoals genactiviteit (transcriptomica) of eiwitten (proteomica). Ze veronderstellen vaak specifieke statistische eigenschappen die niet gelden voor andere datatypen, en ze hebben moeite wanneer het bulkweefsel celtypen bevat die ontbreken in de referentiedata. Sterke batch-effecten—verschillen in donoren, instrumenten of gezondheidsstatussen—kunnen de biologische signalen verder vervagen. Opmerkelijk is dat er geen praktische methode bestond voor metabolomica, de studie van kleine moleculen die vaak het dichtst bij klinische symptomen staan. Wetenschappers die multiomics-cohorten analyseerden, moesten daarom meerdere gespecialiseerde tools hanteren, elk met eigen eigenaardigheden, wat het moeilijk maakte om resultaten tussen studies en datatypen te vergelijken.
Een universele ontwarringsmotor
DECODE pakt deze uitdagingen aan door deconvolutie te behandelen als een flexibel deep-learningprobleem dat genen, eiwitten en metabolieten op een uniforme manier kan verwerken. Eerst synthetiseert het “pseudoweefsels” door single-cell-profielen digitaal in willekeurige verhoudingen te mengen, waardoor een rijk trainingsset ontstaat waarvan de werkelijke celcompositie bekend is. Een adversariële leerfase leert vervolgens een encoder om zowel echte weefsels als pseudoweefsels in een gedeelde representatie te plaatsen waarin technische verschillen worden geminimaliseerd maar biologisch betekenisvolle patronen behouden blijven. Daarna leert een speciaal denoising-module, aangestuurd door contrastief leren, echte weefselsignalen te scheiden van kunstmatige ruis. Deze stap maakt DECODE robuust tegen ontbrekende celtypen in de referentie en tegen meetfouten. Ten slotte worden de opgeschoonde features doorgegeven aan een deconvolutiemodule die afhankelijk van de volledigheid van de referentie absolute of relatieve abundanties van celtypen en celtoestanden schat.
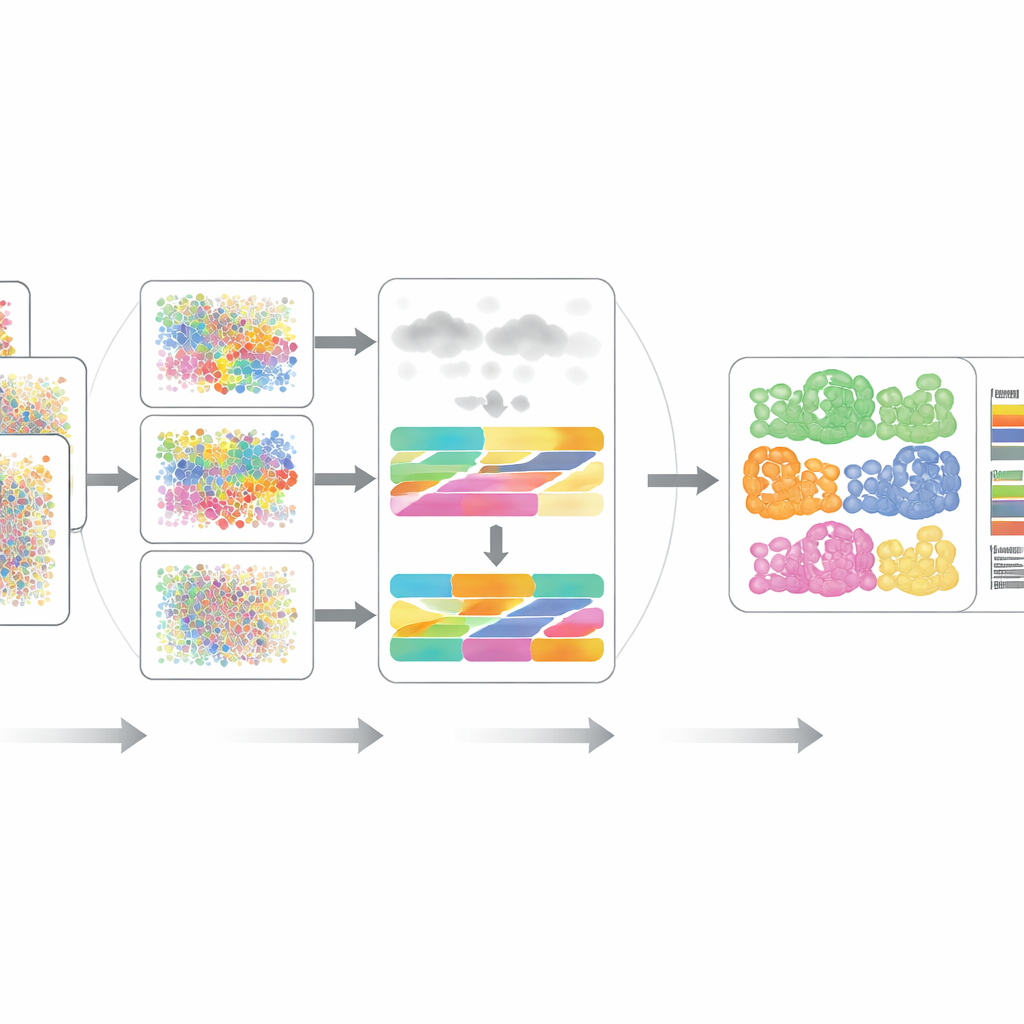
DECODE aan de tand voelen
De auteurs hebben DECODE grondig gebenchmarkt op 15 datasets die zeven realistische scenario’s omvatten, waaronder verschillende donoren, ziekte- en gezondheidscondities, experimentele platformen en zelfs ruimtelijk gerichte metingen. Over transcriptomica en proteomica heen presteerde DECODE over het algemeen gelijk aan of beter dan state-of-the-art tools wat betreft nauwkeurigheid, terwijl de rekentijd en het geheugenverbruik redelijk bleven. Cruciaal is dat DECODE de enige methode was die betrouwbare resultaten opleverde voor metabolomica-data, waar minder features zijn en verschillende celtypen misleidend op elkaar kunnen lijken. Het framework bleek ook bedreven in het volgen van celtoestanden—zoals voortgang langs een ontwikkelingspad, fasen van de celcyclus of reacties op geneesmiddelenbehandeling—in plaats van alleen statische celtypen.
Robuust in rumoerige en onvolledige real-world data
In echte weefsels komen vaak celtypen voor die niet in lab-gebaseerde single-cell-referenties zijn vastgelegd, en experimentele ruis kan veel kenmerken tegelijk vertekenen. De onderzoekers simuleerden deze problemen door onbekende celtypen toe te voegen en verschillende vormen van ruis en missende data te introduceren in transcriptomica, proteomica en metabolomica. In de meeste gevallen bleef DECODE de meest nauwkeurige methode en, in metabolomica, de enige die niet faalde. Ze toonden ook aan dat DECODE zeer consistente antwoorden geeft wanneer het wordt toegepast op overeenkomende gen- en eiwitmetingen van dezelfde bloedcelmonsters, een belangrijke voorwaarde om veranderingen in celtypen over omics-lagen heen in grote cohorts te vergelijken.
Nieuwe biologische inzichten uit multiomics-cohorten
Gewapend met dit eendrachtige hulpmiddel ging het team complexe ziekte-datasets opnieuw langs. Bij borstkanker combineerden ze transcriptomische en proteomische cohorten om te laten zien hoe immuuncellen en ondersteunende stroma-cellen verschuiven tussen niet-metastatische tumoren, metastaserende primaire tumoren en hersenmetastasen. Patronen zoals hogere aantallen T-cellen en perivasculaire-achtige cellen in niet-metastatische laesies en meer B-cellen in gevorderde ziekte sluiten aan bij en breiden eerdere biologische studies uit. In muizenlever integreerde DECODE transcriptomische, proteomische en metabolomische cohorten om te volgen hoe hepatocyten, endotheelcellen en residentiële immuuncellen veranderen onder verschillende diëten en leverziektemodellen, en reproduceerde bekende trends zoals stijgende fracties Kupffer-cellen bij inflammatoire toestanden.
Wat dit vooruit betekent
Voor een algemeen publiek is de kernboodschap dat DECODE werkt als een slimme prisma voor biomedische data: gegeven geblende metingen van weefsels kan het de bijdragen van vele verschillende celtypen en -toestanden scheiden, en het doet dat betrouwbaar over meerdere soorten moleculaire metingen. Dit stelt wetenschappers in staat veel meer informatie uit bestaande multiomics-cohorten en biobanken te halen zonder voor elk project nieuwe single-cell-data te hoeven verzamelen. Hoewel de methode nog steeds afhankelijk is van de kwaliteit en reikwijdte van beschikbare single-cell-referenties, en metabolomica-bronnen beperkt blijven, markeert DECODE een belangrijke stap richting routinematige, celniveau-interpretatie van grootschalige humane studies, met potentiële voordelen voor het begrijpen van ziektemechanismen en het sturen van precisiegeneeskunde.
Bronvermelding: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Trefwoorden: multiomics-deconvolutie, single-cell referentie, deep learning in de biologie, metabolomics-analyse, celtypesamenstelling