Clear Sky Science · nl
Variatie in mutatiesnelheid en terugkerende sequentiefouten in fylogenetica op pandemische schaal
Waarom dit van belang is voor toekomstige uitbraken
Wanneer een nieuw virus zich over de wereld verspreidt, racen wetenschappers om het genetische materiaal te lezen en de stamboom te reconstrueren. Die bomen helpen bij het volgen van hoe varianten ontstaan, hoe snel ze zich verspreiden en of beheersmaatregelen werken. Maar tijdens COVID-19 werden miljoenen SARS‑CoV‑2-genomen zo snel gesequenced dat verborgen fouten en eigenaardigheden in de data het beeld begonnen te vertekenen. Dit artikel introduceert nieuwe methoden om zulke enorme genetische datasets te zuiveren en te interpreteren, en biedt zo helderdere inzichten in hoe een pandemisch virus werkelijk evolueert en zich door populaties verplaatst.
De uitdaging van het begrijpen van miljoenen genomen
Genomische epidemiologie zet virusgenomen om in praktische informatie voor volksgezondheidsbesluiten. Voor SARS‑CoV‑2 zijn wereldwijd meer dan 20 miljoen genomen gedeeld. Traditionele evolutionaire hulpmiddelen zijn ontworpen voor bescheidener vragen, zoals het vergelijken van genen tussen soorten, niet voor het verwerken van miljoenen vrijwel identieke virale sequenties die in real time binnenkomen. Op deze schaal worden twee problemen bijzonder hinderlijk. Ten eerste muteren sommige plaatsen in het virale genoom veel vaker dan andere, waardoor niet-verwante virussen er vreemd vergelijkbaar uit kunnen zien. Ten tweede kunnen terugkerende technische fouten bij sequencing en dataverwerking echte mutaties imiteren. Beide effecten creëren “valse echo’s” in de evolutionaire boom en zorgen voor onzekerheid over welke takken en groeperingen betrouwbaar zijn.
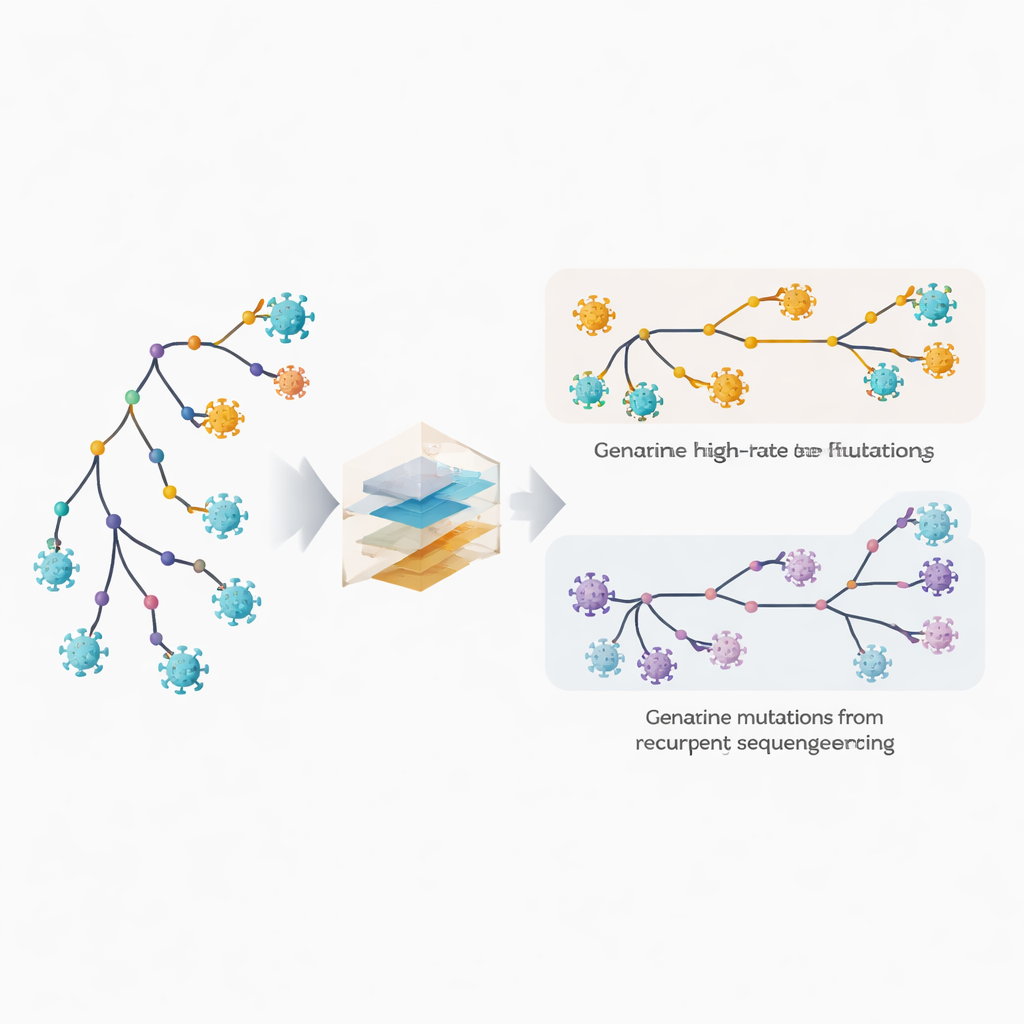
Het opsporen van snel veranderende plaatsen en verborgen fouten
De auteurs breiden hun fylogenetische software MAPLE uit met modellen die elke positie in het virale genoom als een eigen entiteit behandelen. In plaats van uit te gaan van een paar gemiddelde mutatiesnelheden, schat de methode een aparte snelheid voor iedere site, waarbij ze profiteert van het enorme aantal beschikbare genomen. Tegelijkertijd laat het model elke site een eigen kans hebben op het dragen van een terugkerende sequencing- of consensus-bepalingsfout. De sleuteltruc is te vergelijken hoe vaak een verandering voorkomt op diepe interne takken van de boom, die oudere, gedeelde gebeurtenissen weerspiegelen, versus op de buitenste toppen, die overeenkomen met individuele genomen. Echte biologische mutaties zijn meestal verspreid over interne en terminale takken, terwijl technische fouten vooral op de toppen verschijnen. Door dit patroon te benutten kan de methode echte snelle evolutie scheiden van herhaalde fouten.
Snelere algoritmen voor een drukke levensboom
Het verwerken van miljoenen genomen zou normaal gesproken enorme rekenkracht vergen. Om de analyse praktisch te houden, hebben de onderzoekers herontworpen hoe MAPLE sequentie-informatie op de boom opslaat en bijwerkt. In plaats van elk genoom te vergelijken met één vaste referentie, selecteert de software “lokale referentie” punten binnen de boom en legt nearby genomen vast als verschillen ten opzichte van deze ankers. Deze compacte representatie versnelt vergelijkingen tussen verafgelegen delen van de boom. Aanvullende verbeteringen verfijnen hoe nieuwe monsters aan een bestaande boom worden toegevoegd, hoe taklengtes worden afgestemd en hoe waarschijnlijke alternatieve boomvormen worden onderzocht, met opties om de meest veeleisende stappen parallel uit te voeren over meerdere processorkernen.
Het testen van de methode en het opschonen van data uit de praktijk
Om te controleren of hun modellen werken, maakten de auteurs eerst realistische gesimuleerde SARS‑CoV‑2-datasets met bekende mutatiepatronen en ingebedde sequentiefouten. In deze tests herstelde de nieuwe aanpak trouwere evolutionaire bomen en lokalisereerde individuele fouten met hoge precisie, vooral wanneer tienduizenden genomen of meer werden opgenomen. Vervolgens gingen ze naar echte data en analyseerden miljoenen SARS‑CoV‑2-sequenties waarvoor raw reads beschikbaar waren. Door twee verschillende consensusbouw-pijplijnen te vergelijken, identificeerden ze specifieke genoomposities die herhaaldelijk door artefacten werden getroffen, zoals problemen met primerbinding of referentiebias bij het aanroepen. Deze verdachte sites werden gemaskeerd voor verdere analyse, en genomen die tekenen van contaminatie of gemengde infectie vertoonden werden eruit gefilterd, wat resulteerde in een gecureerde uitlijning van meer dan twee miljoen hoogwaardige sequenties.
Een helderder wereldwijd beeld van de virusstamboom
Met de opgeschoonde dataset reconstrueerden de auteurs een mondiale SARS‑CoV‑2-fylogenetische boom en brachten ze in kaart hoe grote varianten zich tot elkaar verhouden. Hun boom stelt soms subtiel verschillende verwantschappen voor dan eerdere openbare bomen, vaak op manieren die minder mutatiegebeurtenissen vereisen en beter bij het statistische model passen. Het raamwerk benadrukt ook waar lijnnaamgeving mogelijk inconsistent is met de onderliggende genetische geschiedenis en markeert mogelijke recombinanten of problematische genomen voor nader onderzoek. Hoewel sommige uitdagingen blijven bestaan—zoals overfitting wanneer data schaars zijn, of de invloed van zwaar verontreinigde monsters—tonen de resultaten aan dat het nu haalbaar is om betrouwbaardere, pandemische-schaal evolutionaire bomen te bouwen. Voor een niet-specialistische lezer komt het erop neer dat beter omgaan met fouten en mutatie-hotspots leidt tot scherpere inzichten in hoe ziekteverwekkers zich verspreiden en veranderen, waardoor wetenschappers en gezondheidsinstanties sneller en met meer vertrouwen kunnen reageren bij toekomstige uitbraken.
Bronvermelding: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Trefwoorden: SARS-CoV-2 genomica, fylogenetische methoden, sequencingfouten, variatie in mutatiesnelheid, genomische epidemiologie