Clear Sky Science · nl
Overdraagbare enantioselectiviteitsmodellen uit schaarse gegevens
Een slimmere manier om de juiste katalysator te vinden
Schemici zoeken vaak naar betere geneesmiddelen en materialen door koolstofatomen op zeer specifieke driedimensionale manieren aan elkaar te koppelen. Het bereiken van dat subtiele „rechts- versus linkshandige” resultaat — bekend als enantioselectiviteit — betekent meestal het uitproberen van veel metaal-katalysatoren en reactiestanden via trial-and-error. Dit artikel introduceert een methode om met relatief kleine hoeveelheden experimentele gegevens, gecombineerd met snelle computerberekeningen, te voorspellen welke nikkelgebaseerde katalysatoren de gewenste handigheid zullen geven in een breed scala aan reacties, waarmee scheikundigen mogelijk weken of maanden aan laboratoriumwerk besparen.
Waarom gehandschoende moleculen zo moeilijk te beheersen zijn
Veel geneesmiddelen en natuurlijke producten bestaan als spiegelfiguurvormen die in het lichaam heel verschillend kunnen gedragen. Katalysatoren die de ene spiegelfiguur boven de andere bevoordelen, zijn daarom zeer waardevol. Maar het ontwerpen van zulke katalysatoren is lastig. Traditionele kwantumchemie kan in principe berekenen welk pad een reactie verkiest, maar kleine energiefouten vertalen zich in grote fouten in de voorspelde selectiviteit, en de berekeningen zijn traag. Simpelere statistische modellen zijn daarentegen snel, maar negeren vaak de gedetailleerde wisselwerking tussen de metaal-katalysator en de reagerende moleculen, vooral wanneer het reactiemechanisme subtiel kan veranderen afhankelijk van de gebruikte partners.
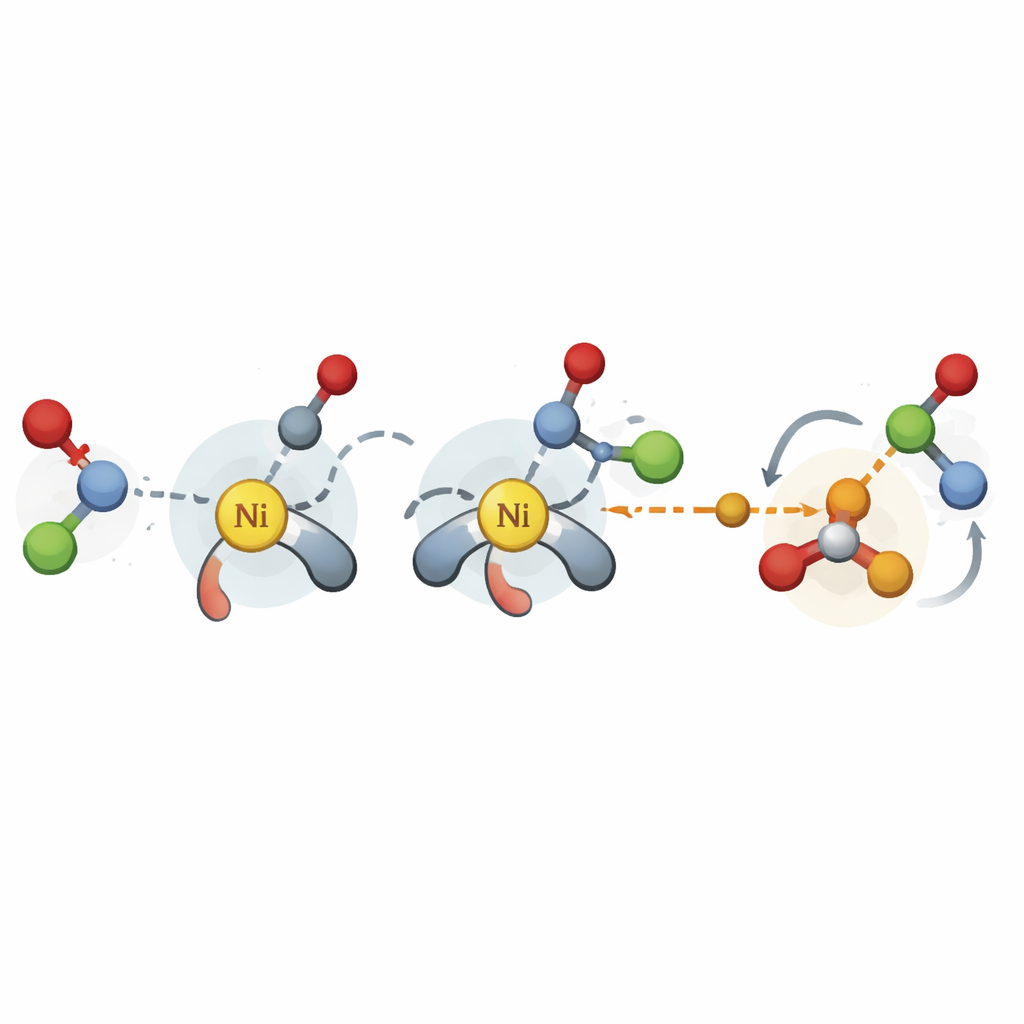
De belangrijke momenten in een reactie vastleggen
De auteurs overbruggen deze kloof door zich te concentreren op de meest cruciale stadia van een nikkelgecatalyseerde kruis-koppelingsreactie: de stappen waarin nieuwe koolstof–koolstofbindingen worden gevormd en het eindproduct wordt losgelaten. In plaats van dure hoog-niveau simulaties uit te voeren, gebruiken ze een vereenvoudigde kwantummethode om driedimensionale structuren te genereren voor sleutelovergangstoestanden en tussenproducten over vele mogelijke katalysator- en substraatcombinaties. Uit deze structuren halen ze honderden fysisch betekenisvolle descriptoren, zoals hoe druk het katalysatormilieu is nabij bepaalde atomen of hoe gemakkelijk elektronen kunnen bewegen. Deze getallen worden vervolgens ingevoerd in eenvoudige lineaire regressiemodellen die structurele eigenschappen koppelen aan de gemeten selectiviteit.
Leren van schaarse gegevens om nieuwe experimenten te sturen
Een centrale prestatie van het werk is dat het het beste haalt uit schaarse gegevens — de beperkte combinaties van katalysatoren en substraten die gewoonlijk in een onderzoeksartikel worden gerapporteerd. In een casestudy bekijkt het team een nikkelreactie die styreenoxiden koppelt met aryljodiden opnieuw. Ze tonen aan dat descriptoren afkomstig van de meest relevante overgangstoestand het beter doen dan die van vereenvoudigde katalysatorfragmenten, zelfs wanneer de onderliggende berekeningen goedkoper zijn. Met deze modellen testen ze virtueel veel meer liganden op bestaande substraatparen en identificeren ze nieuwe katalysatorkeuzes die de enantiomere overmaat verhogen voor bijzonder hardnekkige voorbeelden, terwijl ze tientallen onnodige experimenten vermijden.
Kennis overdragen tussen verschillende reacties
De aanpak is krachtig omdat ze overdraagbaar is naar verschillende, maar gerelateerde, nikkelgecatalyseerde reacties. In een tweede reeks studies combineren de auteurs gegevens van meerdere soorten nikkelreacties die alle bindingen vormen tussen sp3-gehybridiseerde koolstofatomen en partners zoals aryl- of alkenylgroepen, zelfs wanneer de exacte omstandigheden of koppelpartners verschillen. Door modellen te bouwen op basis van dezelfde mechanistisch betekenisvolle descriptoren, voorspellen ze met succes enantioselectiviteit voor nieuwe liganden, nieuwe substraatcombinaties en zelfs een geheel nieuwe klasse van koolstof–koolstofbindingsvorming die niet in de trainingsset was opgenomen. Analyse van welke descriptoren het meest van belang zijn, geeft ook aanwijzingen welke stap in de katalytische cyclus de handigheid bepaalt voor elke reactiefamilie.
Helpen dat scheikundigen sneller nieuwe reacties starten
In een laatste demonstratie gebruiken de auteurs hun descriptorenschema samen met een Bayesian optimalisatieplatform om een nikkelgecatalyseerde koppeling van benzylic acetalen en aryljodiden te ontwerpen die eerder niet asymmetrisch was ontwikkeld. Beginnend met literatuurgegevens over andere reacties, raadt het model kleine batches van veelbelovende liganden aan om te testen, waarmee het in slechts enkele tientallen experimenten snel de best presterende klasse vindt. Voor een scheikundige betekent dit een praktisch hulpmiddel om een nieuw katalytisch project „cold start” te geven: door een handvol vroege resultaten in te voeren, kan het model suggereren welke chirale liganden het meest waarschijnlijk hoge enantioselectiviteit opleveren. Over het geheel genomen toont de studie aan dat zorgvuldig gekozen, goedkope computationele kenmerken beperkte historische gegevens kunnen omzetten in breed bruikbare richtlijnen voor het bouwen van de volgende generatie selectieve reacties.
Bronvermelding: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Trefwoorden: asymmetrische katalyse, nikkel kruis-koppeling, machine learning in chemie, reactie-optimalisatie, enantioselectiviteitsvoorspelling