Clear Sky Science · nl
Graph atomic cluster expansion voor fundamentele machine learning interatomaire potentialen
Computers leren de atomen aanvoelen
Het ontwerpen van nieuwe materialen voor batterijen, vliegtuigen of fusie-reactoren komt vaak neer op een eenvoudige vraag: hoe duwen en trekken atomen aan elkaar? Deze krachten exact berekenen is zo kostbaar dat het voor één materiaal dagen kan kosten op een supercomputer. Dit artikel introduceert een nieuwe familie machine-learningmodellen, GRACE genaamd, die fungeren als een universele "rekenmachine" voor atomaire krachten over het merendeel van het periodiek systeem. Ze zijn bedoeld om nauwkeurige simulaties van complexe materialen routineus in plaats van heroïsch te maken.
Één model voor veel materialen
De meeste bestaande machine-learning krachtvelden zijn specialistische instrumenten: ze werken erg goed voor een paar elementen of verbindingen, maar moeten helemaal opnieuw worden opgebouwd wanneer nieuwe elementen worden toegevoegd. GRACE kiest een andere weg. Het is vanaf het begin ontworpen als een fundamenteel model dat 89 chemische elementen en een enorme verscheidenheid aan atomaire ordeningen met één gedeelde set regels aankan. Hiervoor bouwen de auteurs voort op een wiskundig kader dat de atomic cluster expansion wordt genoemd en breiden dat uit naar grafachtige structuren, waardoor het model zowel lokale atoomomgevingen als meer uitgezette patronen op een verenigde manier kan beschrijven. In plaats van elke mogelijke interactie hard te coderen, leert GRACE compacte "embeddings" die overeenkomsten tussen elementen vastleggen, zodat kennis over het ene materiaal kan helpen bij het beschrijven van een ander.
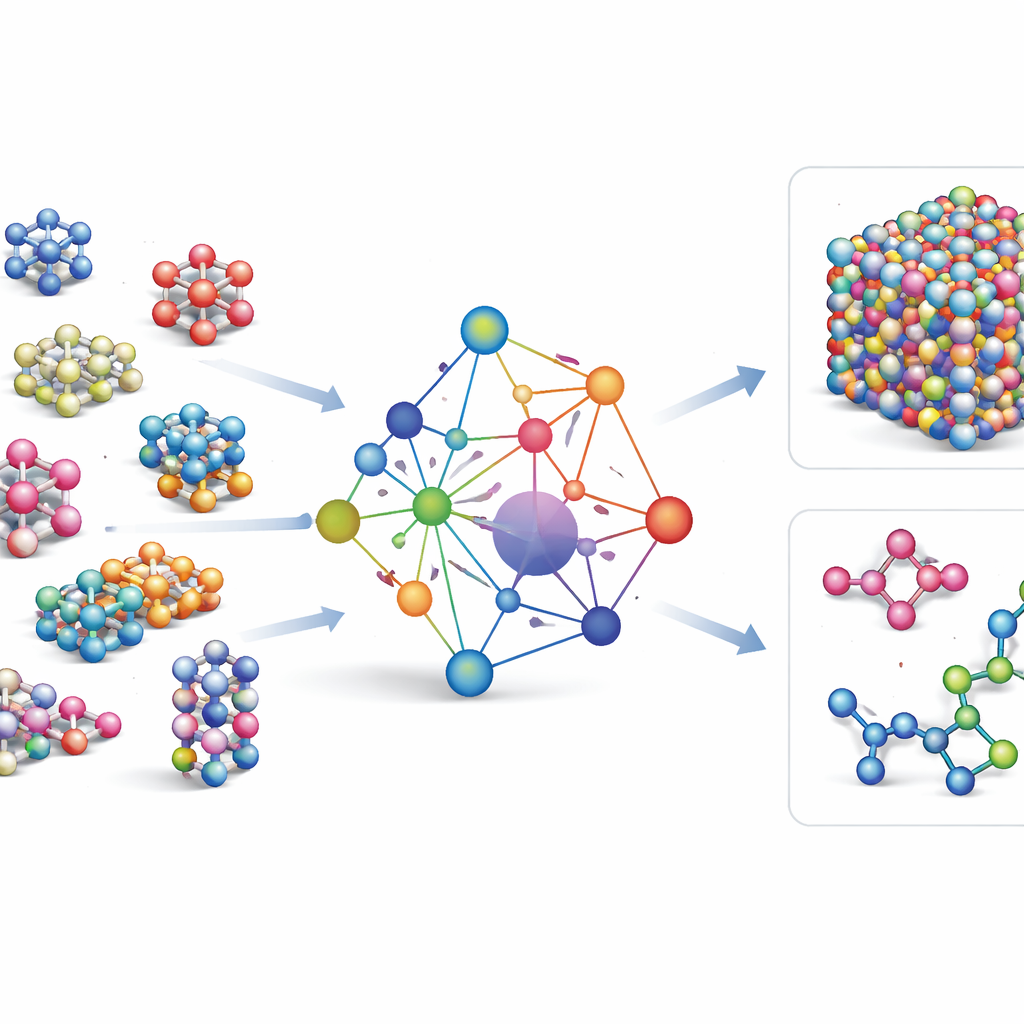
Trainen op een zee van atomaire data
Om GRACE te leren hoe atomen zich gedragen, verzamelden de auteurs enkele van de grootste openbare databanken met kwantummechanische berekeningen. De kern is de OMat24-collectie, die ongeveer 110 miljoen simulaties van anorganische materialen bevat, aangevuld met twee andere die volgen hoe structuren ontspannen en evolueren. Gezamenlijk dekken deze datasets bijna-evenwichtskristallen, uitgerekte en vervormde structuren, hoge-temperatuur vloeistoffen en meer, over dezelfde brede set elementen. GRACE-modellen zijn er in verschillende groottes, van eenvoudigere éénlaagversies die alleen lokale atoomomgevingen bekijken tot diepere tweelaagversies die effectief "berichten" tussen aangrenzende regio's doorgeven. De initiële training streeft naar een goede balans tussen energieën, krachten en interne spanningen, en verdere fijnafstemming past de modellen aan zodat ze compatibel zijn met veelgebruikte referentiedatabanken in de materiaalkunde.
Het model op de proef stellen
Een universeel model is pas nuttig als het betrouwbaar presteert over vele taken. De auteurs onderwerpen GRACE daarom aan een veeleisende testset die weerspiegelt hoe wetenschappers atomaire simulaties daadwerkelijk gebruiken. Op een gemeenschapsbenchmark voor het ontdekken van stabiele kristalstructuren staat GRACE consequent op de "Pareto-front": voor een gegeven nauwkeurigheid is het sneller dan concurrerende modellen, en voor een gegeven snelheid is het nauwkeuriger. Vergelijkbare voordelen verschijnen bij het voorspellen van thermische geleidbaarheid, een eigenschap die scherp afhangt van kleine veranderingen in atomaire bewegingen. GRACE presteert ook goed op elastische eigenschappen, oppervlaktespanningen, korrelgrensenergieën en vormingsenergieën van puntdefecten in veel zuivere metalen, die allemaal onderzoeken hoe materialen reageren op rekken, snijden of lokale schade. Een lange moleculaire-dynamica-run van een heet gesmolten zout toont dat het model numeriek stabiel blijft gedurende nanoseconden terwijl het gedetailleerde structurele patronen en atomaire diffusiesnelheden reproduceert.
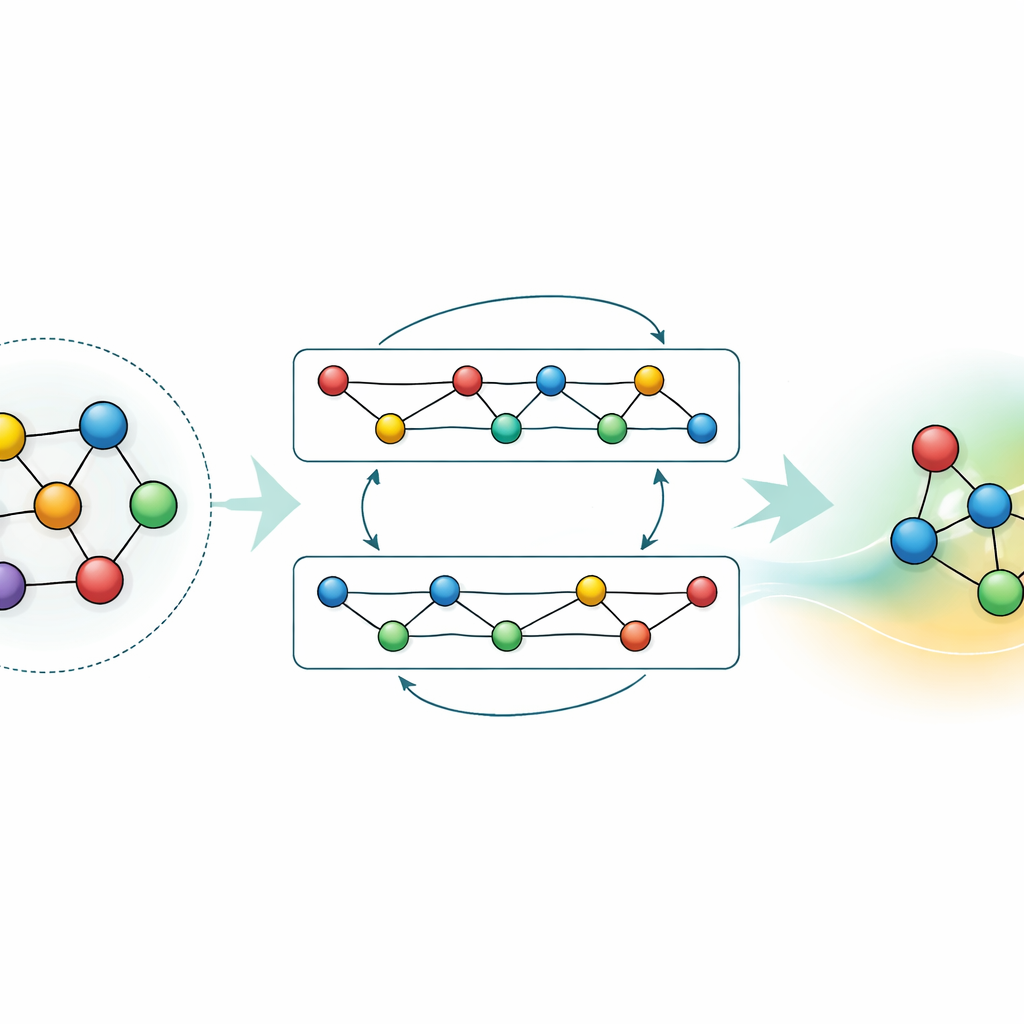
Kennis aanpassen en comprimeren
Hoewel een algemeen model krachtig is, hebben veel toepassingen óf hogere nauwkeurigheid voor een specifiek materiaal óf snellere berekeningen op bescheiden hardware nodig. De auteurs demonstreren twee strategieën om dit te bereiken zonder weg te gooien wat GRACE al heeft geleerd. Ten eerste fijnregelen ze het fundamentele model op gerichte datasets, zoals aluminium–lithium legeringen of gedetailleerde waterstofverbrandingsroutes. Voor de legeringen scherpt zelfs een bescheiden hoeveelheid extra data de voorspellingen aanzienlijk aan, en overtreft modellen die vanaf nul zijn getraind met dezelfde informatie. Voor verbranding zou naive fijnregeling normaal gesproken ertoe leiden dat het model vergeet wat het over andere materialen wist; door zorgvuldig delen van het netwerk te bevriezen en alleen geselecteerde parameters bij te werken, beperken de auteurs dit catastrofale vergeten terwijl ze toch meer nauwkeurigheid voor de nieuwe chemie bereiken. Ten tweede tonen ze aan hoe het grote model kan worden gedistilleerd tot een veel eenvoudiger "student" die de leraar nabootst op sleutelsystemen. Deze gedistilleerde versie draait ongeveer zeventig keer sneller op een CPU en behoudt toch het grootste deel van de nauwkeurigheid, vooral wanneer deze wordt getraind op een mix van complexe legeringen en eenvoudigere referentiestructuren gelabeld door de oorspronkelijke GRACE.
Wat dit betekent voor toekomstig materiaalontwerp
Het werk positioneert GRACE als een flexibel fundament voor de volgende generatie atomaire modellering. In plaats van voor elk materiaal of elke eigenschap een nieuw potentieel te creëren, kunnen onderzoekers starten vanaf een universeel GRACE-model en het vervolgens fijnregelen of distilleren naar hun behoeften, wat enorme hoeveelheden rekentijd en deskundig werk bespaart. De benchmarks tonen aan dat deze aanpak niet alleen bestaande hulpmiddelen evenaart; ze overtreft ze vaak in zowel snelheid als betrouwbaarheid, met name voor veeleisende eigenschappen zoals thermische transport. Voor niet‑specialisten is de kernboodschap dat één goed ontworpen machine-learningmodel nu kan fungeren als een breed vertrouwde "motor" voor virtuele experimenten over een groot deel van het periodiek systeem, waardoor de zoektocht naar betere batterijen, katalysatoren, structurele legeringen en energiematerialen wordt versneld.
Bronvermelding: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Trefwoorden: machine learning interatomaire potentialen, materiaalmodellering, atoomsimulaties, fundamentele modellen, graph atomic cluster expansion