Clear Sky Science · nl
Zelfoptimaliserende machine-learning-potentiaal ondersteunde geautomatiseerde workflow voor zeer efficiënte materiaalkunde van complexe systemen
Slimmere zoektochten naar nieuwe materialen
Het ontwerpen van nieuwe materialen is een beetje alsof je een naald zoekt in een bijna oneindige hooiberg. Van betere batterijen en snellere computers tot efficiëntere laserbronnen en mogelijke supergeleiders bij kamertemperatuur: veel toekomstige technologieën hangen af van het vinden van precies de juiste atomaire ordeningen. Dit artikel beschrijft een manier om kunstmatige intelligentie het grootste deel van die zoektocht automatisch te laten uitvoeren, waardoor de tijd en kosten om veelbelovende nieuwe verbindingen te vinden drastisch verminderen.
Waarom de materiaalpuzzel zo moeilijk is
De eigenschappen van een vaste stof — hoe goed hij elektriciteit geleidt, hoe sterk hij is, hoe hij op licht reageert — worden bepaald door de ordening van zijn atomen in driedimensionale patronen die kristalstructuren worden genoemd. In theorie kun je de kwantummechanica gebruiken om te berekenen welke ordeningen stabiel zijn en wat hun eigenschappen zullen zijn. In de praktijk zijn deze kwantumberekeningen zo belastend dat slechts een klein deel van alle mogelijke materialen kan worden onderzocht. De uitdaging neemt snel toe wanneer meer dan twee chemische elementen betrokken zijn, omdat het aantal combinaties en atomaire ordeningen explodeert, waardoor een blinde zoektocht onuitvoerbaar wordt.
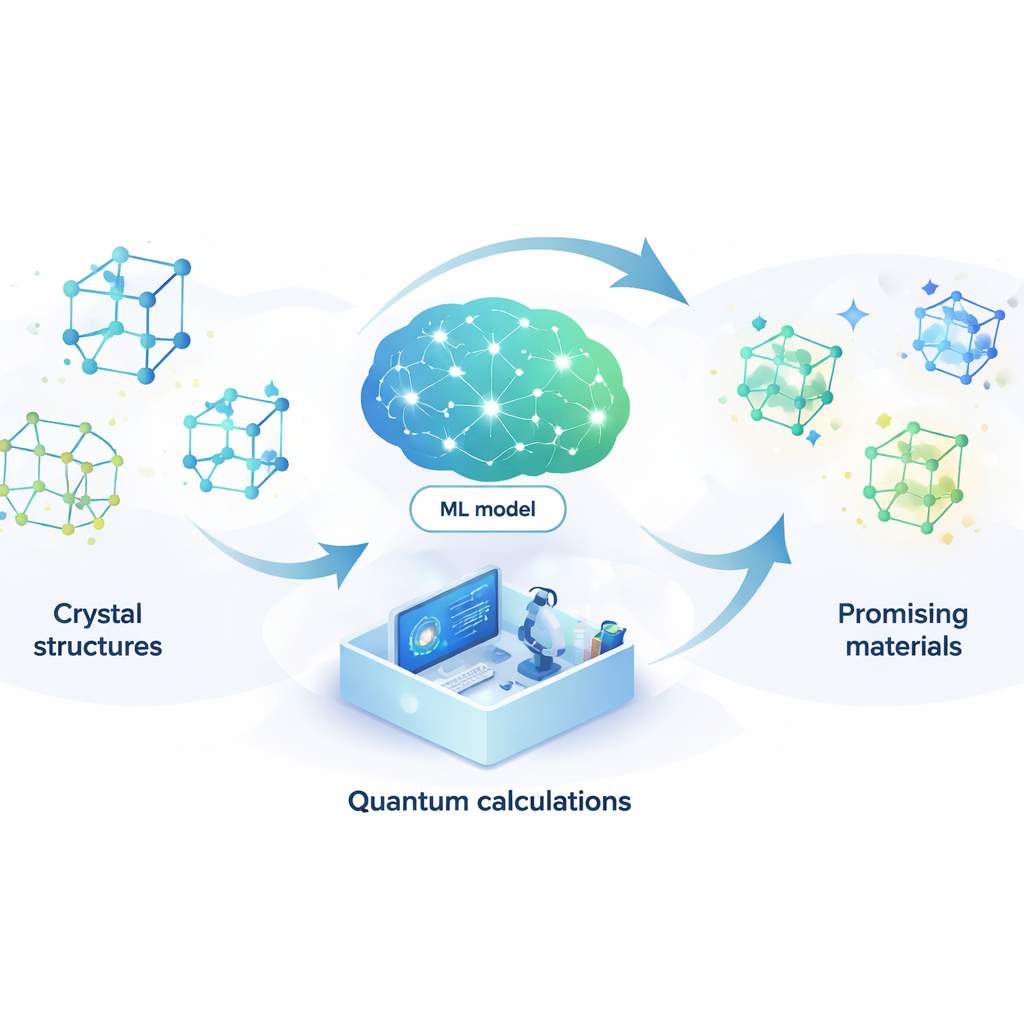
Het laten vervangen van kwantumfysica door een leermodel
Om dit probleem aan te pakken bouwen de auteurs een machine-learningmodel dat de uitkomsten van kostbare kwantumberekeningen kan nadoen tegen een fractie van de kosten. Hun model, een attention-coupled neural network (ACNN), leert hoe de energie van een materiaal afhangt van de posities en types van zijn atomen. Eenmaal getraind kan het zeer snel inschatten of een voorgestelde kristalstructuur waarschijnlijk stabiel is en welke krachten op elk atoom werken. Cruciaal is dat het model zo is ontworpen dat het basisfysische vereisten respecteert, zoals het feit dat het verschuiven of roteren van het gehele kristal de totale energie niet mag veranderen.
Een zelfverbeterende lus voor materiaalontdekking
In plaats van het model één keer te trainen en te hopen dat het overal goed presteert, verpakken de auteurs het in een zelfoptimaliserende lus. Het proces begint met een kleine set willekeurige kristalstructuren, die worden geëvalueerd met volledige kwantummechanische berekeningen en worden gebruikt om een initiële ACNN te trainen. Dit model wordt vervolgens gebruikt om miljoenen proefstructuren te relaxen en snel lokale energieminima te vinden — kandidaatstabiele of bijna-stabiele fasen. De workflow markeert automatisch twee bijzonder waardevolle typen structuren: die welke er erg stabiel uitzien en die welke onfysisch of verdacht lijken. Alleen deze geselecteerde gevallen worden teruggestuurd naar de dure kwantumsolver, en de nieuwe resultaten worden in het model gevoed voor hertraining. Over vele rondes wordt het model gestaag nauwkeuriger in de regio's van de structuurruimte die het meest belangrijk zijn.
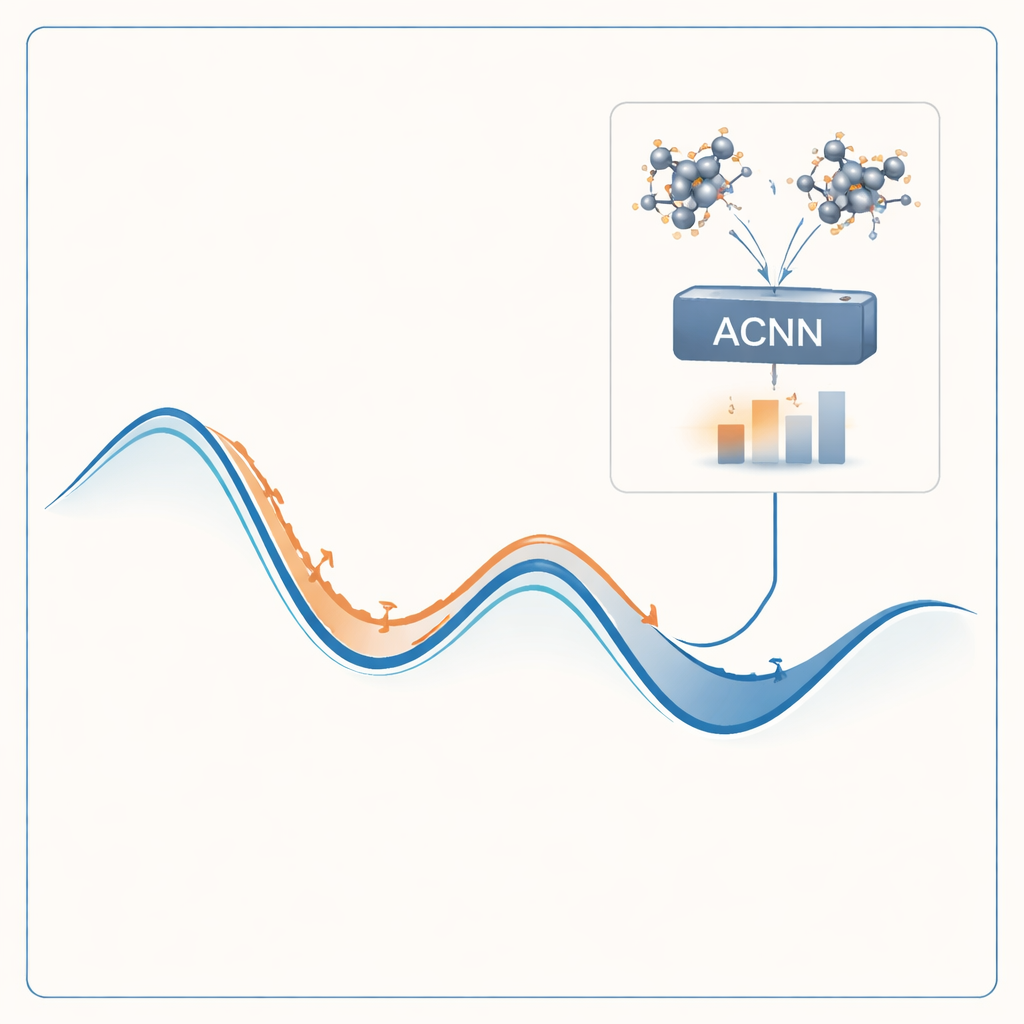
De methode aan de tand gevoeld
Het team demonstreert hun aanpak op twee veeleisende systemen. Het eerste is een hogedrukmengsel van magnesium, calcium en waterstof, een familie van verbindingen van groot belang voor hoogtemperatuursupergeleiding. Door bijna zes miljoen proefstructuren te verkennen, ontdekt hun workflow een nieuwe stabiele fase, MgCa₃H₂₃, en verschillende nauw verwante waterstofrijke “kooi”-structuren. Berekeningen suggereren dat sommige hiervan kunnen supergeleiden bij temperaturen boven het kookpunt van vloeibare stikstof onder extreme druk. De tweede test richt zich op een vier-elementensysteem met beryllium, fosfor, stikstof en zuurstof, gekozen vanwege het potentieel voor kristallen die laserlicht efficiënt kunnen omzetten naar diep-ultraviolette golflengten. Hier relaxeert de methode meer dan negen miljoen structuren en identificeert drie thermodynamisch stabiele fasen met zeer brede bandbanden en veelbelovende optische eigenschappen.
Van brute kracht naar gerichte ontdekking
In beide voorbeelden behaalt de geautomatiseerde workflow snelheidsverbeteringen van ongeveer tienduizend keer vergeleken met het uitsluitend gebruik van kwantumberekeningen, terwijl hij toch betrouwbaar structuren aanwijst die nader onderzoek verdienen. Voor een niet-specialist is de kernboodschap dat een groot deel van materiaalontdekking nu kan worden afgehandeld door een leersysteem dat zichzelf vertelt waar het onzeker is en alleen gerichte, hoogprecisie-berekeningen vraagt wanneer dat nodig is. Dit soort zelfcorrigerende, AI-ondersteunde zoektocht maakt het mogelijk veel complexere mengsels van elementen te verkennen dan voorheen haalbaar was, en vergroot de kans nieuwe supergeleiders, optische kristallen en andere functionele materialen te vinden die de volgende generatie technologieën ondersteunen.
Bronvermelding: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Trefwoorden: materiaalontdekking, machine-learning-potentiëlen, kristalstructuurvoorspelling, supergeleidende hydrides, niet-lineaire optische kristallen