Clear Sky Science · nl
DiNovo maakt hoog-dekkende en hoog-vertrouwenswaardige de novo-peptidevolgordebepaling mogelijk via mirror-proteases en deep learning
Proteïnen in nieuw detail zichtbaar maken
Proteïnen zijn de microscopische machines die onze cellen in leven houden, maar de volledige samenstelling van hun bouwstenen uitlezen blijft verrassend lastig. Dit artikel introduceert DiNovo, een nieuw softwaresysteem dat onderzoekers helpt proteïnefragmenten veel vollediger en betrouwbaarder te "lezen" dan voorheen. Door een slimme biochemische truc te combineren met moderne kunstmatige intelligentie belooft het verborgen eiwitten, ziekte-indicatoren en zelfs immuun-doelen te onthullen die traditionele methoden vaak missen.
Waarom het lezen van proteïnefragmenten zo moeilijk is
De meeste eiwitanalyse van vandaag berust op het in kleinere stukjes snijden van eiwitten, zogenaamde peptides, en vervolgens het wegen van hun fragmenten in een massaspectrometer. Vanuit die gewichten proberen computers de oorspronkelijke peptidevolgorde te reconstrueren, als het oplossen van een kruiswoordraadsel met gedeeltelijke aanwijzingen. Bestaande methoden veronderstellen meestal dat peptiden uit bekende proteïne-databases komen, wat goed werkt voor vertrouwde eiwitten maar worstelt met nieuwe of onverwachte varianten. Zogenaamde de novo-volgordebepaling omzeilt deze beperking door te proberen peptiden rechtstreeks uit de data te lezen, maar slaagt daar vaak niet in omdat sommige fragmenten ontbreken en sommige peptiden nooit schoon worden geknipt.
Mirror-enzymen gebruiken om de gaten op te vullen
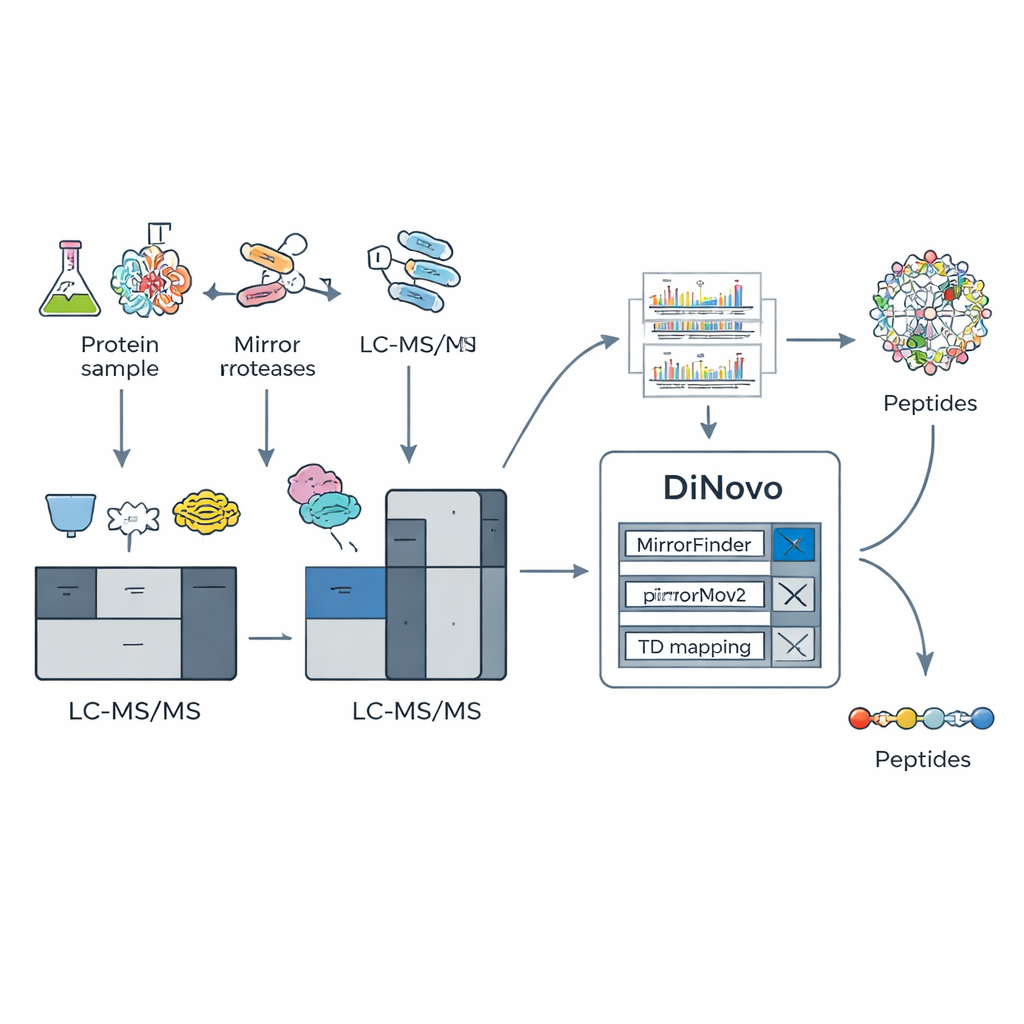
Het kernidee achter DiNovo is het gebruik van paren “mirror-proteases” – paren knipenzymen die eiwitten aan tegenovergestelde zijden van hetzelfde type aminozuur doorsnijden. Bijvoorbeeld: het ene enzym knipt nét voor een lysine, terwijl het partnerenzym nét na die lysine knipt. Dit levert twee gerelateerde peptiden op die hetzelfde binnenste segment delen maar verschillende uiteinden hebben. Wanneer deze "mirror"-peptiden worden geanalyseerd, bevatten hun massaspectra elkaar aanvullende fragmentpatronen: wat in het ene spectrum ontbreekt, verschijnt vaak in het andere. De auteurs tonen aan dat het combineren van zulke mirror-paren de fragmentdekking tot bijna compleet kan opdrijven, met ongeveer 98% van mogelijke knipplaatsen ondersteund door echte experimentele signalen — veel hoger dan wanneer slechts één enzym wordt gebruikt.
Een slimme softwarepijplijn gebouwd voor mirror-data
Om deze biochemische truc te benutten, bouwde het team DiNovo als een end-to-end softwareworkflow. Eerst worden eiwitten van bacteriën en gist verteerd met twee mirror-paren enzymen, en worden de resulterende peptiden geanalyseerd met hoge-resolutie massaspectrometrie. DiNovo gebruikt vervolgens een module genaamd MirrorFinder om automatisch te herkennen welke spectra-paren van mirror-peptiden afkomstig zijn, waarbij dit direct uit signaalpatronen gebeurt in plaats van uit eerdere sequentieaanwijzingen. Daarna gebruikt de hoofd de novo-engine MirrorNovo deep learning om die gepaarde spectra te interpreteren, terwijl een secundaire grafgebaseerde engine, pNovoM2, een snellere CPU-only optie biedt. Samen vertalen deze tools pieken naar aminozuurvolgordes en onderzoeken ze ook individuele spectra die geen duidelijke paren vormden, om zoveel mogelijk informatie uit de data te persen.
Vertrouwen meten zonder te steunen op oude databases
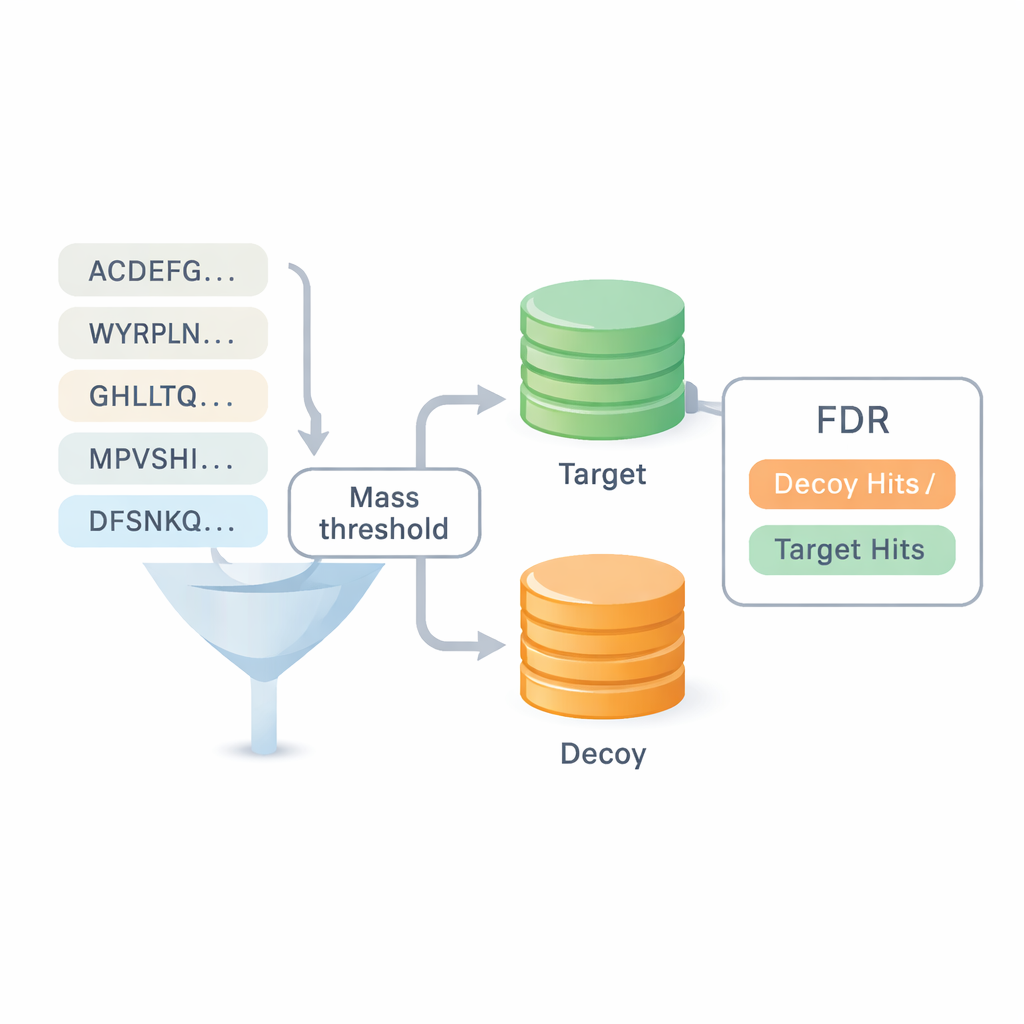
Een van de grootste vragen bij de novo-volgordebepaling is hoeveel vertrouwen je in resultaten kunt stellen. De meeste bestaande benchmarks hergebruiken antwoorden uit databasezoekopdrachten, wat de grens tussen de twee benaderingen vervaagt en fouten kan verbergen. DiNovo introduceert een andere kwaliteitscontrolemethode genaamd target-decoy mapping. Hierbij worden de nieuw gelezen peptiden gemapt op een gecombineerde verzameling van echte (target) en kunstmatig, door elkaar gehusselde (decoy) proteïnevolgordes. Door te vergelijken hoe vaak peptiden in de echte set versus de gehusselde set terechtkomen, kan de software een foutpercentage, of false discovery rate, schatten zonder te leunen op eerdere identificaties. Dit maakt het mogelijk DiNovo rechtstreeks te vergelijken met standaard database-zoekprogramma's onder dezelfde foutcontroles.
Wat DiNovo in de praktijk oplevert
In tests op bacteriële, gist- en antilichaammonsters las DiNovo consequent veel meer peptiden en aminozuren dan bekende de novo-tools die slechts één enzym gebruiken. Met twee mirror-paren produceerde het 2–3 keer meer hoog-vertrouwde aminozuren dan een klassieke trypsineenmalige opstelling en identificeerde het meer eiwitten bij vergelijkbare foutniveaus. In directe vergelijkingen met drie toonaangevende database-zoekengines vond DiNovo vergelijkbare aantallen aminozuren en eiwitten, en de meeste van zijn sequenties kwamen overeen met die van de zoekengines op dezelfde spectra. De auteurs stellen dat dit niveau van dekking en overeenstemming betekent dat de novo-volgordebepaling, lang behandeld als een reservemethode, nu naast databasezoekopdrachten kan staan als een serieuze en in sommige gevallen superieure optie.
Grote lijn: richting volledige, onbevooroordeelde eiwitlezing
Voor niet-specialisten is de conclusie dat DiNovo het veel eenvoudiger maakt om proteïnefragmenten nauwkeurig te lezen zonder beperkt te zijn tot wat al in referentiedatabases staat. Door de hoeveelheid goed-onderbouwde sequentie-informatie te verdubbelen of verdrievoudigen en door ingebouwde foutcontroles te bieden, opent deze benadering de deur naar het ontdekken van onbekende eiwitten, het volgen van subtiele variaties en het verkennen van complexe mengsels waarin veel componenten nog onduidelijk zijn. Kortom, door mirror-enzymen te koppelen aan deep learning en zorgvuldige statistiek, helpt DiNovo ruwe spectrale sporen om te zetten in een helderder, betrouwbaarder beeld van de eiwitten die ten grondslag liggen aan gezondheid en ziekte.
Bronvermelding: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Trefwoorden: proteomica, de novo-peptidevolgordebepaling, massaspectrometrie, deep learning, mirror-proteases