Clear Sky Science · nl
Efficiënte bemonstering van grootschalige overgangspaden en tussenliggende conformaties in sub-mesoscopische proteïnecomplexen
Proteïnen in beweging bekijken
Veel van de moleculen die ons in leven houden gedragen zich minder als stijve Lego-blokjes en meer als kleine machines die voortdurend van vorm veranderen. Deze bewegingen drijven processen aan zoals energieproductie, DNA-reparatie en het binnendringen van virussen in cellen. Experimenten zoals cryo-elektronenmicroscopie kunnen nu enkele van deze structuren ‘invriezen’, maar niet de vluchtige tussenstappen. Dit artikel introduceert eBDIMS2, een nieuwe rekenmethode die de ontbrekende frames van proteïnebeweging kan vullen, zelfs voor enorme moleculaire machines die voorheen te groot en te complex waren om op een gewone computer te simuleren.
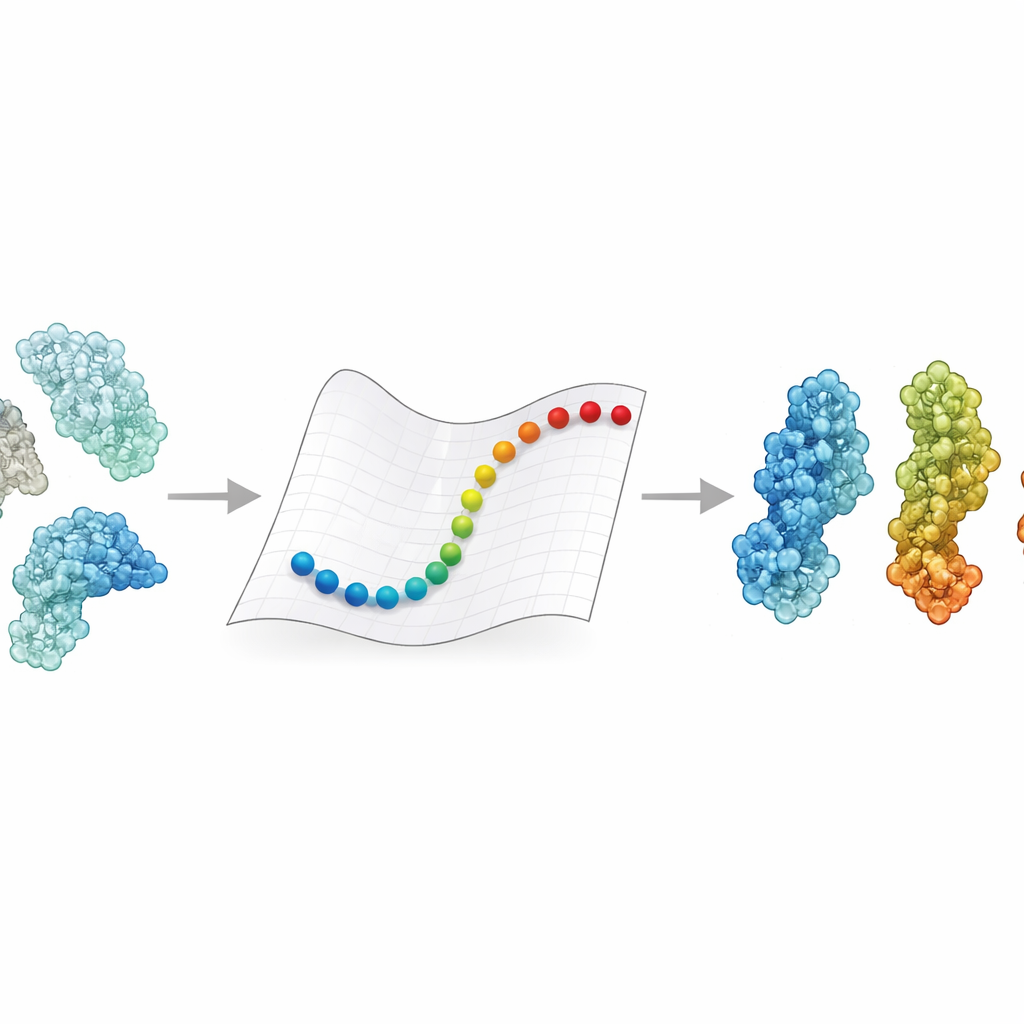
Waarom veranderingen in eiwitvorm belangrijk zijn
Proteïnen blijven zelden in één houding bevroren. Ze openen en sluiten, draaien en buigen als reactie op signalen zoals veranderende spanningen, pH of het binden van een partnermolecuul. Deze veranderingen kunnen het verschil betekenen tussen een enzym dat actief of inactief is, of een receptor die een virus vangt of laat ontsnappen. Experimenten geven gedetailleerde momentopnames van een paar sleutelformaten, en moleculaire dynamica-simulaties kunnen in principe die vormen met elkaar verbinden door elk atoom in de tijd te volgen. Maar het volgen van dergelijke beweging voor de enorme complexen die nu met cryo-elektronenmicroscopie worden waargenomen — vaak met massa’s van honderdduizenden tot miljoenen Dalton — vereist meestal supercomputers en weken rekenwerk. Daardoor weten we voor veel medisch belangrijke reuzen nog steeds niet hoe de ene toestand in de andere overgaat.
Een snellere route door proteïnelandschappen
eBDIMS2 neemt een afkorting door te vereenvoudigen hoe eiwitten worden voorgesteld en hoe hun beweging wordt berekend. In plaats van elk atoom te volgen, behandelt het elke aminozuurresidu als een enkel punt dat verbonden is met veren in een elastisch netwerk. Deze veren vangen op hoe verschillende delen van het eiwit de neiging hebben samen te bewegen. De methode gebruikt vervolgens Brownse dynamica — wiskundige regels die het schudden in een vloeistof nabootsen — om de structuur van een experimenteel bekende toestand naar een andere toe te duwen. Cruciaal is dat eBDIMS2 alleen aandacht besteedt aan interacties die echt van belang zijn, met afstandsafkappunten en parallelle berekening om de kosten te verminderen. Dit verbetert de schaalbaarheid van de software van ruwweg kwadratisch naar bijna lineair met de grootte van het eiwit. In de praktijk betekent dit dat overgangen voor enorme assemblages van bijna twee miljoen Dalton in enkele uren op een desktop kunnen worden verkend, in plaats van praktisch onbereikbaar te zijn.
De paden toetsen aan echte eiwitten
Om te beoordelen of deze snelle paden biologisch zinnig zijn, stelden de auteurs ensembles samen van 47 grote eiwitten en 15 aanvullende complexen, in totaal honderden structuren die grotendeels met cryo-elektronenmicroscopie waren bepaald. Ze gebruikten hoofdcomponentenanalyse, een statistisch gereedschap dat de dominante bewegingswijzen van elk eiwit identificeert, om deze structuren te ordenen in conformationele landschappen zoals open, gesloten, actief of inactief. eBDIMS2 werd vervolgens gevraagd paren eindtoestanden door dit landschap te verbinden. De resulterende paden werden teruggeprojecteerd op dezelfde laag-dimensionale kaarten, om te laten zien of ze vloeiende routes volgen die dicht langs experimenteel waargenomen tussenstappen lopen. In meer dan 30% van de systemen liepen de gesimuleerde routes dichtbij — binnen enkele ångström — van tussenliggende structuren die niet als input waren meegegeven. Voor veeleisende gevallen zoals het DNA-reparatie-enzym DNA-PKcs of het coronavirus spike-eiwit, overlosten de coarse-grained paden ook goed met veel duurdere atoomniveau-simulaties, waaronder gerichte moleculaire dynamica en geavanceerde enhanced-sampling runs.
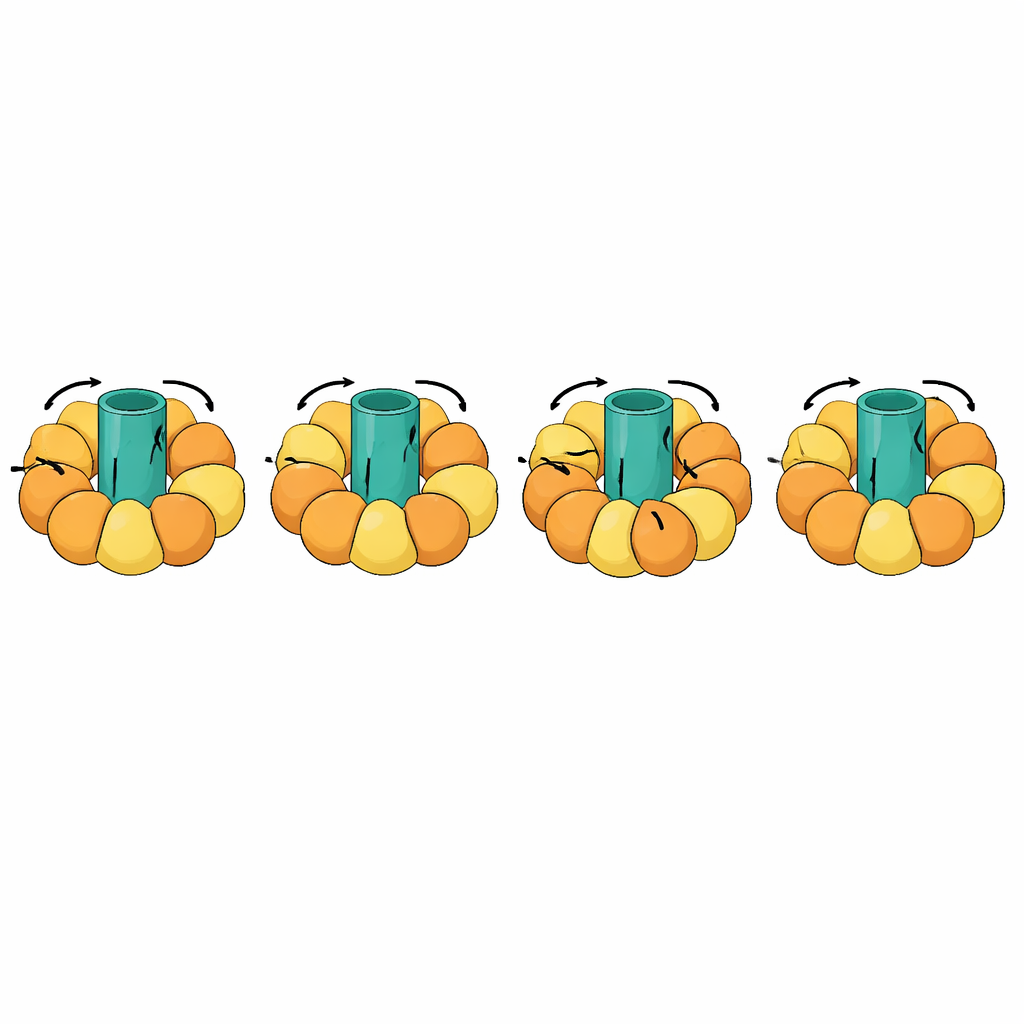
Grote moleculaire machines volgen
Een van de meest opvallende tests betrof roterende machines zoals ATP-synthases, die de energievaluta van de cel maken door een draaiende rotor in het membraan te koppelen aan open- en sluitbewegingen in omringende subeenheden. Deze overgangen zijn buitengewoon complex: delen van het molecuul moeten stijf blijven en als een eenheid roteren, terwijl andere delen in een gechoreografeerde cyclus buigen. eBDIMS2 introduceert speciale behandeling voor dergelijke quasi stijve stukken en voor onvolledige experimentele modellen met ontbrekende segmenten, beide veelvoorkomend in cryo-elektronenmicroscopie. Met deze functies kan het volledige rotatiecycli van ATP-synthase en andere massieve complexen zoals moleculaire chaperonnes, receptoren en virale assemblages simuleren. Gedurende dit proces vermijden de gegenereerde tussenstructuren de ernstige vervormingen die door sommige concurrerende methoden worden geproduceerd en kunnen ze worden opgeschoond tot atomistische modellen die geschikt zijn voor geneesmiddelontwerpberekeningen of langere, meer gedetailleerde simulaties.
Wat dit betekent voor biologie en geneeskunde
De studie toont aan dat eBDIMS2 op betrouwbare wijze de belangrijkste routes tussen bekende eiwitvormen kan schetsen voor systemen die voor traditionele simulaties buiten bereik waren. Het vervangt geen gedetailleerde atoomniveaufilms of levert precieze energieën en tijdschalen, maar het biedt een snelle, fysisch gefundeerde manier om in kaart te brengen hoe grote moleculaire machines zich mogelijk bewegen, met slechts een paar experimentele structuren als input. Naarmate structurele databanken zich vullen met meerdere toestanden van grote eiwitassemblages die verbonden zijn met kanker, infectie en andere ziekten, geeft deze benadering onderzoekers een toegankelijk instrument om de verbanden te leggen, plausibele tussenstaten voor te stellen en te wijzen waar te zoeken met hogere-resolutiemethoden of gericht geneesmiddelontwerp.
Bronvermelding: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Trefwoorden: proteïnedynamica, moleculaire simulaties, cryo-EM, conformationele paden, coarse-grained modellering