Clear Sky Science · nl
Activering van IRF3 in cardiomyocyten schaadt de mitochondriale oxidatieve functie via remming van PGC-1α en veroorzaakt hartfalen
Waarom gestreste harten en vermoeide cellen ertoe doen
Hartfalen wordt vaak beschreven als het ‘slijten’ van het hart, maar onder de motorkap is het ook een verhaal over chronische ontsteking en uitgeputte energiecentrales binnen hartspiercellen. Deze studie stelt een ogenschijnlijk eenvoudige vraag met grote implicaties: bestaat er één moleculaire schakelaar in hartcellen die schadelijke ontsteking en falende energieproductie verbindt — en zo ja, kan het omzetten van die schakelaar het beloop van hartfalen veranderen? Door die draad te volgen, brengen de auteurs een sleutelspeler aan het licht en tonen zij aan dat het voorzichtig versterken van het eigen energieprogramma van het hart deels falende harten bij muizen kan herstellen.
Een moleculaire schakelaar in zieke menselijke harten
De onderzoekers richtten zich op een eiwit genaamd IRF3, vooral bekend om zijn rol bij de cellulaire reactie op virale infecties. Ze bestudeerden weefsel van mensen met ischemische cardiomyopathie, een veelvoorkomende vorm van hartfalen veroorzaakt door verminderde bloedtoevoer na hartinfarcten. In deze falende harten was IRF3 niet alleen aanwezig — het was chemisch geactiveerd op specifieke plaatsen, een aanwijzing dat het actief genprogramma’s aanstuurt. Tegelijkertijd was de machinerie waarmee mitochondriën brandstof omzetten in energie via oxidatieve fosforylering duidelijk verzwakt. Een vergelijkbaar patroon verscheen in muismodellen van hartinfarct: wanneer een kransslagader werd afgeklemd, raakte IRF3 in hartspiercellen sterk geactiveerd en gingen IRF3-gestuurde genen aan. Zelfs fragmenten van mitochondriaal DNA — vrijgekomen uit beschadigde mitochondriën en fungerend als interne ‘gevaar-signalen’ — waren voldoende om IRF3 in geïsoleerde hartcellen te activeren.
IRF3 uitschakelen beschermt het hart
Om te testen of IRF3-activiteit in hartspiercellen de ziekte daadwerkelijk verergert, maakten de onderzoekers muizen waarin IRF3 alleen uit cardiomyocyten kon worden verwijderd, terwijl andere immuun- en ondersteunende cellen intact bleven. Na het veroorzaken van een hartinfarct hadden deze muizen een betere pompfunctie en minder littekenvorming dan normale muizen, ondanks een gelijke initiële verwonding. In hartcellen die in kweek werden gekweekt, dempte het uitschakelen van IRF3 ontstekingsgenen zonder andere verwante eiwitten te verstoren. Gezamenlijk wijzen deze resultaten erop dat IRF3 binnen de hartcel zelf niet slechts een bijstander is: het versterkt ontsteking en structurele schade na ischemie en draagt bij aan de overgang naar hartfalen.
Als IRF3 “aan” vastzit, stort het brandstofsysteem in
De auteurs keerden het experiment om: ze creëerden muizen waarin IRF3 in cardiomyocyten gedwongen in een permanent actieve staat kon worden gezet met een slimme genetische ‘fosfomimetische’ truc. Zelfs zonder extern prikkel ontwikkelden deze muizen snel ernstige hartdysfunctie, hoge niveaus van ontstekingsmediatoren in het bloed en tekenen van celschade. Een diepgaande analyse van hun hartweefsel toonde aan dat chronische IRF3-activiteit een meesterregulator van energie, PGC-1α, onderdrukt. Deze molecule bevordert normaal gesproken gezonde mitochondriën, efficiënte verbranding van vetten en gebalanceerde cellulaire energie. Met PGC-1α naar beneden gedrukt, daalden meerdere mitochondriale eiwitten, stokte de elektronentransportketen en veranderden de brandstofkeuzes van het hart: carnitine en aanverwante verbindingen voor vetverbranding namen af, ketongebruik was verstoord en glucoseverwerking raakte ontregeld. Zelfs de verhouding NAD⁺/NADH — een belangrijke indicator van het cellulaire redoxevenwicht — schoof in de verkeerde richting.
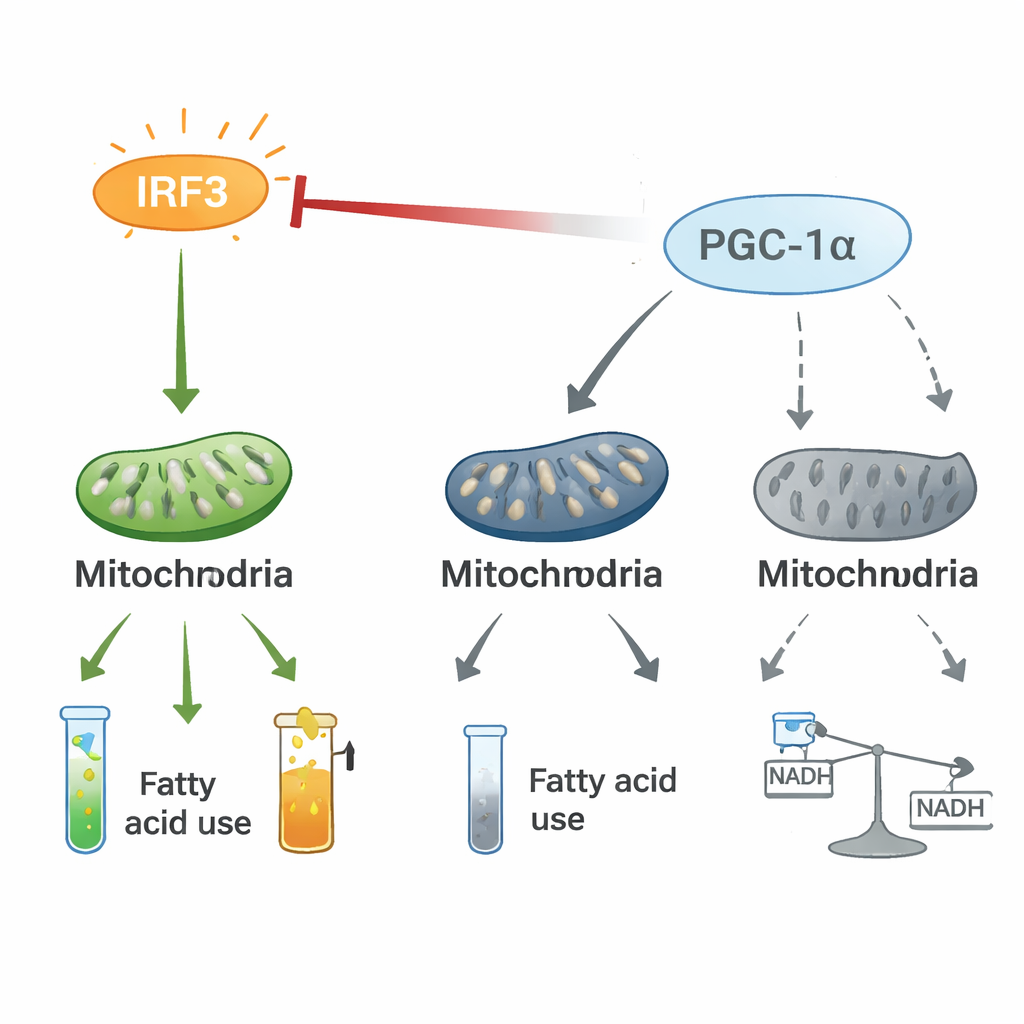
Een touwtrekken tussen ontsteking en energieregulatie
Mechanistische experimenten lieten zien dat IRF3 en PGC-1α een tweerichtingsregulerende as vormen. In hartcellen associeert geactiveerd IRF3 fysiek met PGC-1α en vermindert het diens vermogen om vetverbrandingsgenen aan te zetten. Het onderdrukken van IRF3 verhoogt PGC-1α-niveaus en -activiteit, terwijl het verhogen van PGC-1α IRF3-gedreven ontstekingsgenen dempt en mitochondriale markers herstelt, zelfs onder stressomstandigheden zoals lage zuurstof of bacteriële toxines. Met behulp van stabiele isotopen volgden de auteurs dat IRF3-activatie koolstofstromen wegstuurt van normale energieproductie via de citroenzuurcyclus naar een alternatieve route, de pentosefosfaatroute, en de soepele doorstroom van metabolieten verstoort. Dit touwtrekken tussen een pro-inflammatoire schakelaar (IRF3) en een energiemedepiloot (PGC-1α) lijkt het hartmetabolisme te herschikken op manieren die ontsteking en energieverlies bevorderen.
Het voorzichtig heropladen van de batterijen van het hart
Tot slot vroegen de onderzoekers of het licht verhogen van PGC-1α de schade door IRF3 kon tegengaan. Ze gebruikten een hartgericht gentherapievector om PGC-1α matig — maar niet excessief — te verhogen in dezelfde muizen met hyperactief IRF3. Deze bescheiden verhoging verbeterde de pompfunctie, verhoogde mitochondriale eiwitten, versterkte genen voor vetverbranding en NAD-metabolisme, en verminderde ontstekings- en fibrotische genactiviteit. In celexperimenten herstelde co-expressie van PGC-1α met actief IRF3 een gezondere NAD⁺/NADH-balans en verschuiving van brandstofverbruik terug richting vetten. Voor de niet-specialist betekent dit dat het zorgvuldig heropladen van het ‘batterijbeheersysteem’ van het hart de schadelijke effecten van een chronische inflammatoire schakelaar die op “aan” vastzit gedeeltelijk kan compenseren.
Wat dit betekent voor toekomstige zorg bij hartfalen
Dit werk plaatst IRF3 als een centrale schakel tussen ontsteking en energiefalen binnen hartspiercellen. In plaats van ontsteking en metabolisme als gescheiden problemen bij hartfalen te behandelen, suggereert de studie dat ze met elkaar verweven zijn via een IRF3–PGC-1α-as. Hoewel deze bevindingen in muizen en cellen zijn gedaan, werpen ze de mogelijkheid op dat toekomstige therapieën ofwel IRF3-activiteit kunnen afremmen of PGC-1α en mitochondriale functie kunnen versterken om hartfalen na een infarct te vertragen of te voorkomen. In eenvoudige termen: het kalmeren van een overactief cellulair alarmeringssysteem en het ondersteunen van de energiecentrales van het hart kunnen samen een krachtige strategie blijken om verzwakte harten langer sterk te laten kloppen.
Bronvermelding: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Trefwoorden: hartfalen, ontsteking, mitochondriën, cardiomyocyten, PGC-1α