Clear Sky Science · nl
Een generieke referentie gedefinieerd door consensus-pieken voor single-cell ATAC-seq-gegevensanalyse
Waarom het in kaart brengen van de open deuren van ons DNA ertoe doet
Elke cel in je lichaam draagt in wezen hetzelfde DNA, maar hersencellen, bloedcellen en tumorcellen gedragen zich heel verschillend. Een belangrijke reden is dat slechts bepaalde gedeelten van het DNA op elk moment blootliggen en "open" zijn voor gebruik. Nieuwe single-cell-technologieën kunnen deze openheid nu genoombreed meten, maar tot nu toe ontbrak een gemeenschappelijke referentiekaart—iets als een standaardatlas—om resultaten tussen experimenten en laboratoria te vergelijken. Deze studie bouwt zo’n kaart, genaamd cPeaks, en laat zien hoe die ons beeld van celtypen, ontwikkeling en kanker kan verscherpen.
Veel experimenten omzetten in één gedeelde kaart
De auteurs begonnen met het verzamelen van 624 hoogwaardige experimenten die open chromatine—de toegankelijke delen van het DNA—maatregelen in meer dan 40 menselijke organen. In elk experiment hadden computerprogramma's al "pieken" gemarkeerd waar het DNA bijzonder blootlag. In plaats van elk dataset afzonderlijk te behandelen, legde het team al deze piek-lijsten zorgvuldig langs het genoom over elkaar en voegde overlappende gebieden samen. Ze onderzochten vervolgens hoe vaak elke kleine positie binnen deze samengevoegde regio's in de verschillende experimenten als open werd gemarkeerd, waardoor elke regio een karakteristieke vorm kreeg die weerspiegelde hoe consistent ze voorkomt. Wanneer een samengevoegd gebied meerdere dicht bij elkaar gelegen open plekken bevatte, splitsten ze het in meerdere eenvoudigere eenheden. Deze eenheden—ongeveer 1,4 miljoen in totaal—werden de geobserveerde consensus-pieken, of cPeaks, een kandidaat-referentiecatalogus voor menselijke chromatine-toegankelijkheid. 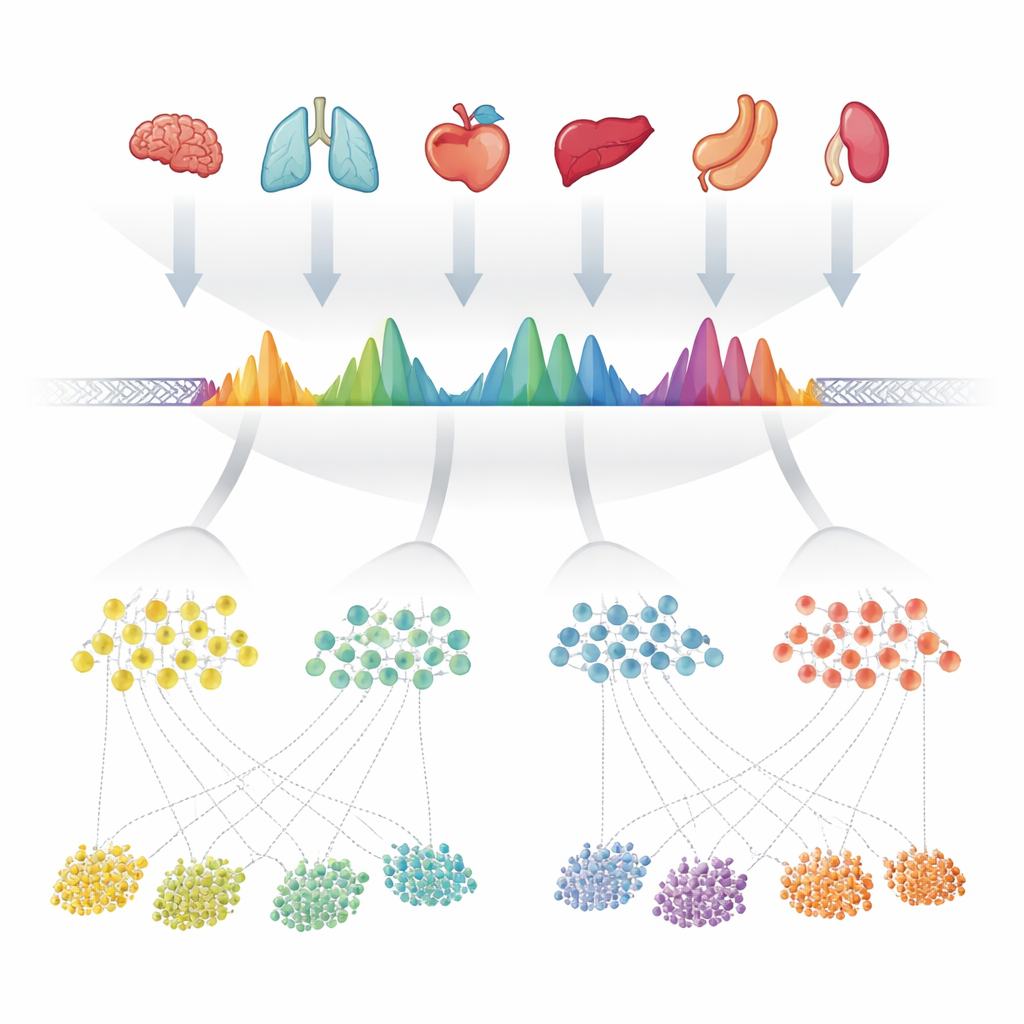
Een stabiel vingerafdruk over weefsels en technologieën
Om een nuttige referentie te zijn, moeten deze cPeaks echte, herhaalbare kenmerken van het genoom vertegenwoordigen, geen eigenaardigheden van specifieke monsters of software. De auteurs testten dit door hun samengevoegde regio's opnieuw te reconstrueren met alleen bloedmonsters, alleen vaste weefsels, afzonderlijke openbare databases en zelfs verschillende laboratoriummethoden om open DNA te onderzoeken. In elk geval leverden dezelfde genomische locaties opmerkelijk vergelijkbare piekvormen op, en overlappen de meeste single-cell-datasets die ze onderzochten meer dan 90% van hun eigen pieken met de cPeak-catalogus. Reads uit veel organen stapelden zich precies op rond cPeak-centrums, wat aantoont dat deze regio's betrouwbaar vastleggen waar chromatine open is. Vergeleken met eerdere referentiesets op basis van gerelateerde technologieën, besloegen cPeaks meer van het toegankelijke DNA dat door ATAC-seq-experimenten werd opgepikt, en vingen ze bijna evenveel signaal op als pieken die vers in elke dataset werden gedefinieerd—ondanks dat ze vast en herbruikbaar zijn.
Een neuraal netwerk leren ontbrekende regio's te vinden
Zelfs honderden bestaande monsters kunnen niet elk mogelijk celtype dekken. Om hun kaart uit te breiden naar regio's die nog niet waren waargenomen, wendde het team zich tot deep learning. Ze trainden een eendimensionaal convolutioneel neuraal netwerk op DNA-sequenties: voorbeelden die binnen geobserveerde cPeaks lagen dienden als positieve voorbeelden, terwijl willekeurig gekozen achtergrondregio's als negatieve voorbeelden fungeerden. Het model leerde deze twee met hoge nauwkeurigheid te onderscheiden, wat impliceert dat cPeaks herkenbare sequentiepatronen dragen. Toen de onderzoekers doelbewust pieken die specifiek waren voor één weefsel per keer verborgen, herstelde het netwerk ze nog steeds op basis van sequentie alleen, inclusief zeldzame weefsel-specifieke plekken. Ze schoven vervolgens een klein venster over de rest van het genoom, keurden elk segment en voegden ongeveer 280.000 hoog scorende nieuwe regio's toe aan de catalogus als voorspelde cPeaks, waarmee de dekking vooral verbeterde in weefsels die in de oorspronkelijke gegevens ondervertegenwoordigd waren.
Open regio's koppelen aan genen, celtypen en zeldzame cellen
Met een rijkere referentie in handen vroegen de auteurs zich af wat deze regio's doen. Veel cPeaks liggen in de buurt van genstart- en eindplaatsen of overlappen bekende regulatorische elementen zoals promotoren, enhancers en bindingsplaatsen voor architecturale eiwitten zoals CTCF. Een kleine subset is in bijna elke dataset toegankelijk; deze langere "housekeeping"-cPeaks bevinden zich vaak in kernpromotorregio's van genen die nodig zijn voor basaal celonderhoud. Het team classificeerde cPeaks ook naar hoe scherp en consistent hun randen zijn over monsters heen, wat weerspiegelt hoe precies het nabije DNA in nucleosomen is verpakt. Regio's met scherp gedefinieerde grenzen zijn verrijkt voor bepaalde families van transcriptiefactoren die bekendstaan om chromatine te hervormen en ontwikkeling aan te sturen. Wanneer cPeaks werden gebruikt als de featureset om meerdere single-cell-datasets te analyseren, verbeterden ze de nauwkeurigheid van celtype-annotatie en waren ze bijzonder nuttig bij het identificeren van zeldzame celtypen en subtiele subtypen die eerdere pieksets of eenvoudige genomische rasters vaak samenvaagden.
Ontwikkeling en kanker volgen met een gemeenschappelijke taal
De kracht van een standaardreferentie wordt duidelijk bij het vergelijken van zeer verschillende biologische contexten. Met cPeaks heranaliseerden de auteurs single-cell-gegevens van het zich ontwikkelende menselijke netvlies, grote atlassen van foetale en volwassen weefsels en meerdere kankersoorten. Ze konden ontwikkelingsbanen reconstrueren en zagen dat het aandeel scherp begrensde, "goed gepositioneerde" cPeaks de neiging heeft toe te nemen tijdens overgangsfases en daarna af te nemen naarmate cellen zich vestigen in stabiele identiteiten. Een vergelijkbaar patroon verscheen over tumorstadia: intermediaire kankers vertoonden een hoger aandeel van deze gestructureerde regio's, wat wees op intense regulatorische herschikking. In één eierstuktumor hielpen cPeaks twee verschillende kankersubklonen met verschillende DNA-kopieaantalveranderingen te onthullen, wat aantoont hoe de referentie verborgen complexiteit in ziekte kan blootleggen.
Wat dit betekent voor toekomstig genoomonderzoek
Voor niet-specialisten kunnen cPeaks worden opgevat als een gestandaardiseerde set coördinaten die aangeven waar het genoom het waarschijnlijkst fysiek open en actief is in veel menselijke celtypen. Door nieuwe single-cell chromatine-experimenten op deze gedeelde kaart uit te lijnen, kunnen onderzoekers resultaten tussen studies vergelijken, zeldzame of overgangsceltoestanden gemakkelijker opsporen en beginnen met het bouwen van grootschalige modellen van genregulatie—vergelijkbaar met hoe gestandaardiseerde gen-catalogi de opkomst van single-cell RNA-atlassen mogelijk maakten. De huidige cPeak-catalogus is een eerste concept dat zal groeien naarmate nieuwe data binnenkomen, maar het biedt al een gemeenschappelijke taal om chromatine-toegankelijkheid te beschrijven en brengt ons dichter bij een verenigd beeld van hoe DNA-verpakking ontwikkeling, gezondheid en ziekte stuurt. 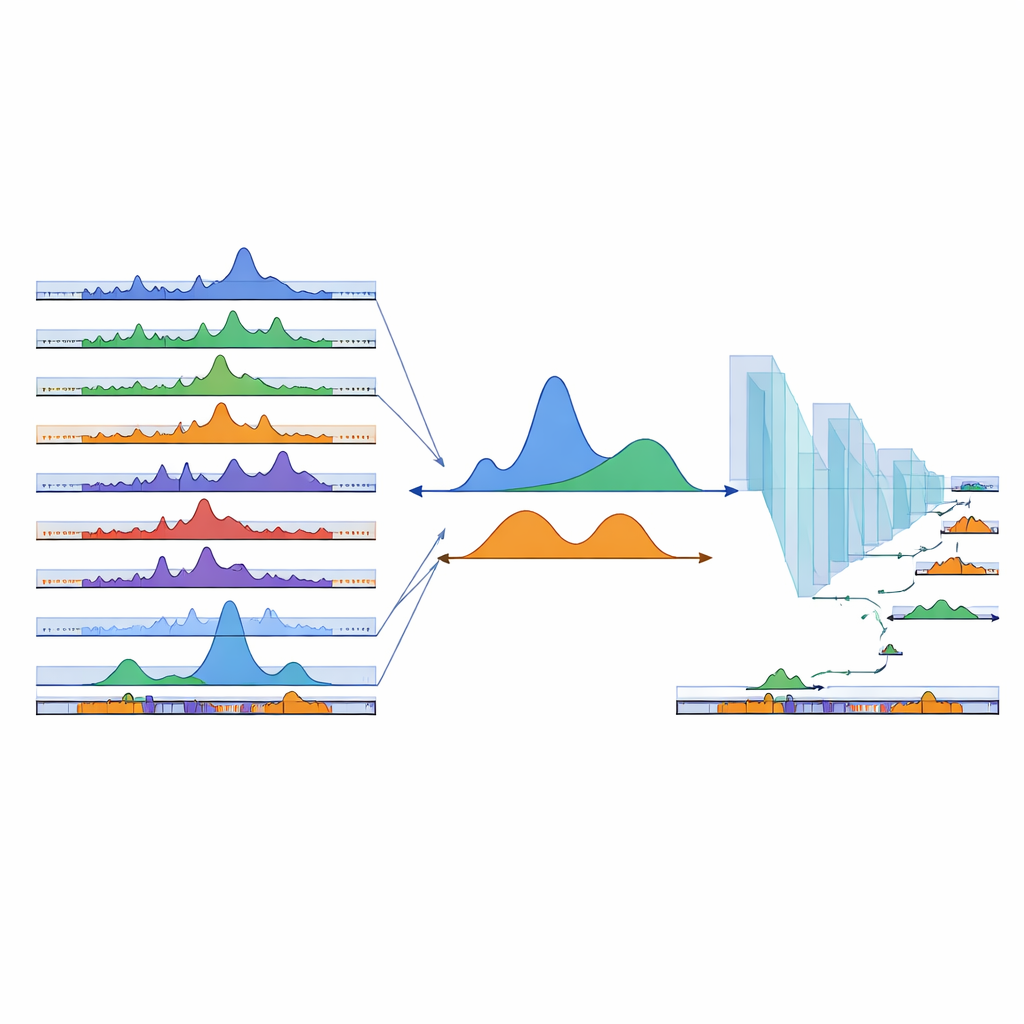
Bronvermelding: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Trefwoorden: chromatine toegankelijkheid, single-cell ATAC-seq, consensus-pieken, genregulatie, deep learning genomica