Clear Sky Science · nl
scLong: een foundation-model met miljarden parameters voor het vastleggen van langafstand-gencontext in single-cell-transcriptomics
Computers leren de verborgen taal van cellen lezen
Elke cel in je lichaam herbergt als het ware een drukke stad van genen die complex aan- en uitgaan. Moderne single-cell RNA-sequencing kan nu elk afzonderlijk exemplaar beluisteren, maar de uitkomst is een overweldigende stroom cijfers. Dit artikel introduceert scLong, een omvangrijk kunstmatig-intelligentie-model dat is ontworpen om deze ingewikkelde patronen van genactiviteit te doorgronden, inclusief zwakke signalen die oudere methoden vaak negeren. Het doel is onderzoekers te helpen begrijpen hoe cellen reageren wanneer genen worden uitgeschakeld, medicijnen worden toegevoegd of ziekten zich ontwikkelen.
Waarom genkaarten op celniveau ertoe doen
Traditionele genstudies mengen vaak miljoenen cellen, waardoor zeldzame of afwijkende cellen worden uitgemiddeld. Single-cell-technieken hebben dat veranderd door genactiviteit per cel afzonderlijk te meten, waardoor verborgen celtypen, subtiele cell-tot-celcommunicatie en gedetailleerde regelkringen zichtbaar worden die bepalen wat een cel doet. Het analyseren van dit soort data is echter uiterst uitdagend: elke cel kan activiteitsniveaus hebben voor tienduizenden genen, waarvan vele nauwelijks detecteerbaar zijn. Bestaande AI-modellen vereenvoudigen het probleem door zich te concentreren op alleen de luidste genen, wat de berekening versnelt maar veel subtiele signalen mist die cruciaal kunnen zijn bij ziekte, ontwikkeling of medicijnrespons.
Een nieuw model dat naar elk gen luistert
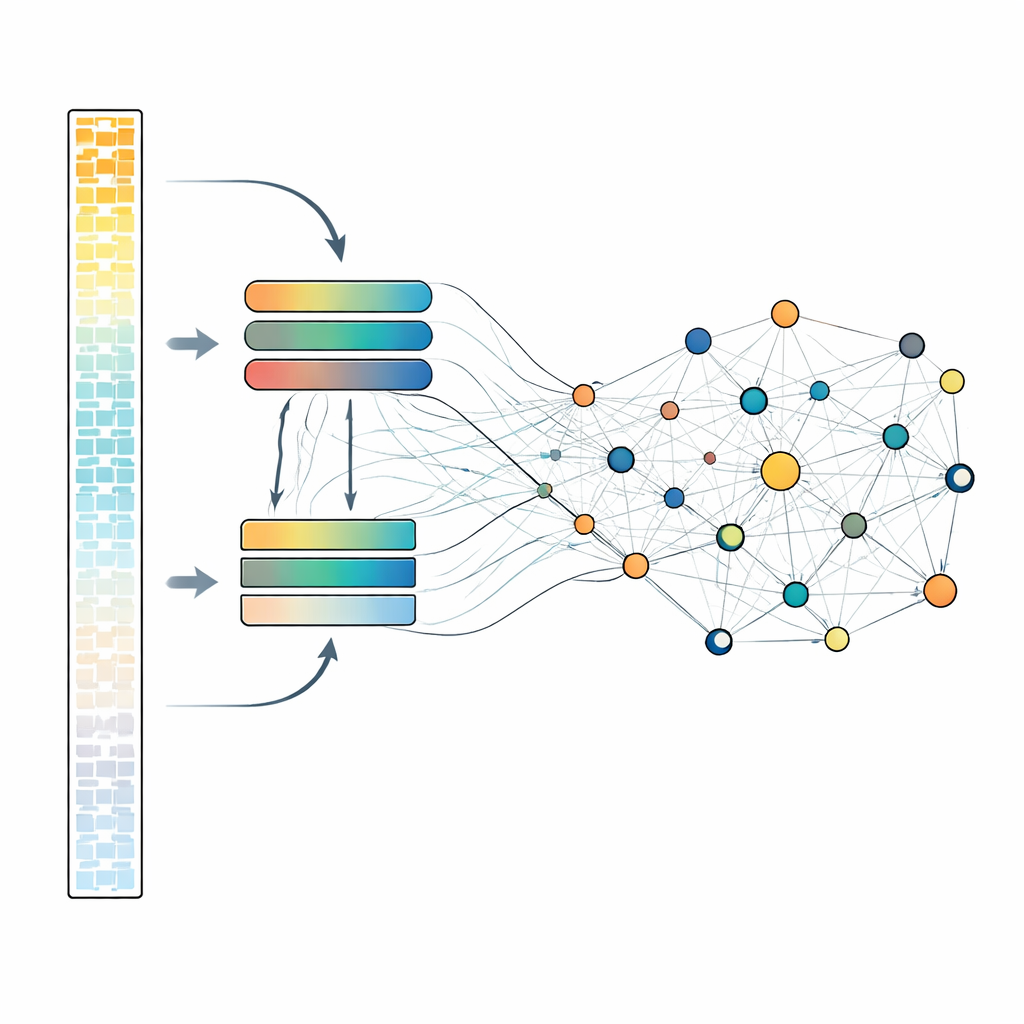
scLong pakt deze uitdaging aan door uit te schalen in plaats van te reduceren. Het is een foundation-model met miljarden parameters, getraind op genactiviteitsprofielen van ongeveer 48 miljoen humane cellen uit meer dan 50 weefsels. In tegenstelling tot vroegere benaderingen die aandacht besteden aan een paar duizend sterk actieve genen, beschouwt scLong ruwweg 28.000 genen tegelijk, inclusief zelden of zwak tot expressie gebrachte genen. Het combineert twee soorten informatie voor elk gen: hoe actief het is in een gegeven cel en wat al bekend is over zijn functie vanuit de Gene Ontology, een groot, deskundig gecureerd register van genrollen en -relaties. Een gespecialiseerd netwerk dat opereert op een graaf van genverbindingen destilleert deze voorkennis tot compacte representaties die het model kan gebruiken naast de ruwe expressiewaarden.
Hoe het model macht en efficiëntie in balans brengt
Elk gen in detail bekijken is rekenkundig kostbaar, dus gebruikt scLong een slim twee-sporenontwerp. Binnen elke cel worden genen gesorteerd op basis van hun expressiesterkte. De meest actieve genen, die vaak het belangrijkste biologische signaal dragen, worden verwerkt via een groter, krachtiger attentie‑module. De stillere genen, inclusief lage en zelfs nulmetingen, worden geleid door een kleinere, lichtere module. Daarna worden alle genen weer samengebracht en door een andere attentielaag geleid die elk gen toestaat elke andere te beïnvloeden. Dit ontwerp maakt het mogelijk om goedkopere maar toch betekenisvolle representaties voor zwakke signalen te behouden, terwijl meer capaciteit is gereserveerd voor de sterkste signalen. Tijdens pretraining verbergt het systeem herhaaldelijk een subset van genactiviteitswaarden en leert die te reconstrueren vanuit de omringende context, waardoor het gedwongen wordt de patronen te ontdekken die genen met elkaar verbinden.
Het model toepassen op echte problemen
Eens getraind kan scLong worden aangepast aan een breed scala aan biologische vraagstukken. De auteurs tonen aan dat het voorspelt hoe genactiviteit verandert wanneer specifieke genen worden uitgeschakeld of aangepast, inclusief combinaties van twee genen die samen kunnen werken. Het voorspelt ook hoe cellen reageren wanneer ze worden blootgesteld aan verschillende chemicaliën, wat belangrijk is voor medicijnontdekking en veiligheidstesten. In kankerstudies helpt scLong te anticiperen op hoe tumorcellen reageren op individuele medicijnen en op koppels van medicijnen die in combinatie effectiever kunnen zijn, vaak beter presterend dan zowel gespecialiseerde modellen als andere grote foundation-modellen. Naast voorspelling kan scLong netwerken van regulatorische relaties tussen genen afleiden en helpen technische vertekeningen te corrigeren die ontstaan wanneer data in verschillende laboratoria of op verschillende apparaten worden verzameld.
Wat dit betekent voor toekomstige geneeskunde en onderzoek
In eenvoudige termen geeft scLong wetenschappers een kaart van genactiviteit binnen individuele cellen met hoge resolutie en contextbewustzijn, een kaart die de stille of zelden gebruikte genen niet weggooit. Door te leren van miljoenen cellen en bestaande biologische kennis te integreren, biedt het nauwkeurigere inschattingen van hoe cellen zullen reageren wanneer genen verstoord worden, wanneer nieuwe medicijnen worden geïntroduceerd of wanneer ziektemechanismen zich ontvouwen. Dit kan de zoektocht naar nieuwe therapieën versnellen, persoonlijkere behandelingskeuzes sturen en ons begrip scherper maken van hoe complexe gennetwerken gezondheid en ziekte beheersen. Hoewel het model groot en rekenkundig veeleisend is, wijst het op een toekomst waarin krachtige, algemene AI-systemen dienstdoen als veelzijdige metgezellen bij het verkennen van de verborgen werking van onze cellen.
Bronvermelding: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Trefwoorden: single-cell transcriptomics, foundation-modellen, genregulatie, voorspelling van medicijnrespons, genexpressie