Clear Sky Science · nl
Het ontsluiten van Cas9-PAM-diversiteit door metagenomische winning en machine learning
Waarom dit belangrijk is voor toekomstige genbewerking
CRISPR is het symbool van moderne genbewerking geworden, maar één stilzwijgende regel beperkt nog steeds wat het kan: elke knip in DNA moet naast een korte “toestemmingsbrief” liggen. Deze korte patronen, PAMs genoemd, bepalen waar het veelgebruikte enzym Cas9 wel of niet kan werken. Deze studie laat zien hoe het doorzoeken van enorme hoeveelheden microbieel DNA, gecombineerd met geavanceerde machine learning, een enorme verborgen variatie aan deze toestemmingsbriefjes kan onthullen. Die nieuwe kaart zou veel meer locaties in het menselijk genoom kunnen openen voor preciezere, veiligere therapieën.
Verborgen regels die CRISPR-knippen sturen
Cas9 en verwante enzymen maken deel uit van een natuurlijk immuunsysteem dat voorkomt bij bacteriën en archaea. Om te voorkomen dat ze hun eigen DNA knippen, zoeken deze microben met Cas-eiwitten naar een PAM—een zeer korte reeks letters—naast de doellocatie. Alleen wanneer die PAM aanwezig is, zal Cas9 het DNA ontwinden en zijn geleide-RNA een match laten controleren, wat een knip veroorzaakt als alles overeenkomt. Het probleem voor de geneeskunde is dat veelgebruikte laboratoriumwerkpaarden, zoals de standaard Cas9 van Streptococcus pyogenes, slechts smalle PAM-patronen herkennen. Als een ziekteveroorzakende mutatie niet het juiste nabije sequentie heeft, kunnen de huidige hulpmiddelen er simpelweg niet bij zonder nauwkeurigheid op te geven.
Het microbieel leven afzoeken voor nieuwe opties
De auteurs wilden systematisch in kaart brengen hoe verschillende Cas9-eiwitten verschillende PAMs in de natuur herkennen. Ze doorzochten meer dan 3,8 miljoen bacteriële en archaeale genomen en meer dan 7,4 miljoen virale en plasmide-sequenties die microben infecteren of tussen hen bewegen. Door CRISPR-arrays te identificeren, deze te koppelen aan nabijgelegen Cas9-genen en vervolgens de opgeslagen “geheugen”-spacers te matchen met binnendringende virussen en plasmiden, konden ze zien welke korte DNA-patronen vaak echte doelen flankeren. Daarmee bouwden ze CRISPR-PAMdb, een openbare catalogus met 8003 Cas9-groepen, elk gekoppeld aan een consensus PAM-profiel, en georganiseerd in een evolutionaire boom die laat zien hoe nauwer verwante Cas9-enzymen vaak vergelijkbare PAM-voorkeuren delen, terwijl ze toch opvallende algehele diversiteit tonen.
Wanneer data opraken, laat het model leren
Zelfs met dit enorme onderzoek ontbrak het bij de meeste gevonden Cas9-eiwitten aan voldoende overeenkomende virale doelen om direct een PAM af te lezen. Om de gaten op te vullen, bouwde het team een machine learning-model genaamd CICERO. CICERO gebruikt een krachtig eiwit-"taalmodel" dat algemene patronen van aminozuursequenties heeft geleerd en verfijnt dit om voor elk gegeven Cas9-eiwit te voorspellen hoe waarschijnlijk het is dat elke DNA-letter op elk van tien posities in de PAM voorkomt. Het model is getraind op de PAM-profielen uit CRISPR-PAMdb en vervolgens getest via kruisvalidering en op 79 Cas9-enzymen waarvan de PAMs experimenteel waren gemeten, waarbij sterke overeenstemming tussen voorspelling en werkelijkheid werd bereikt.
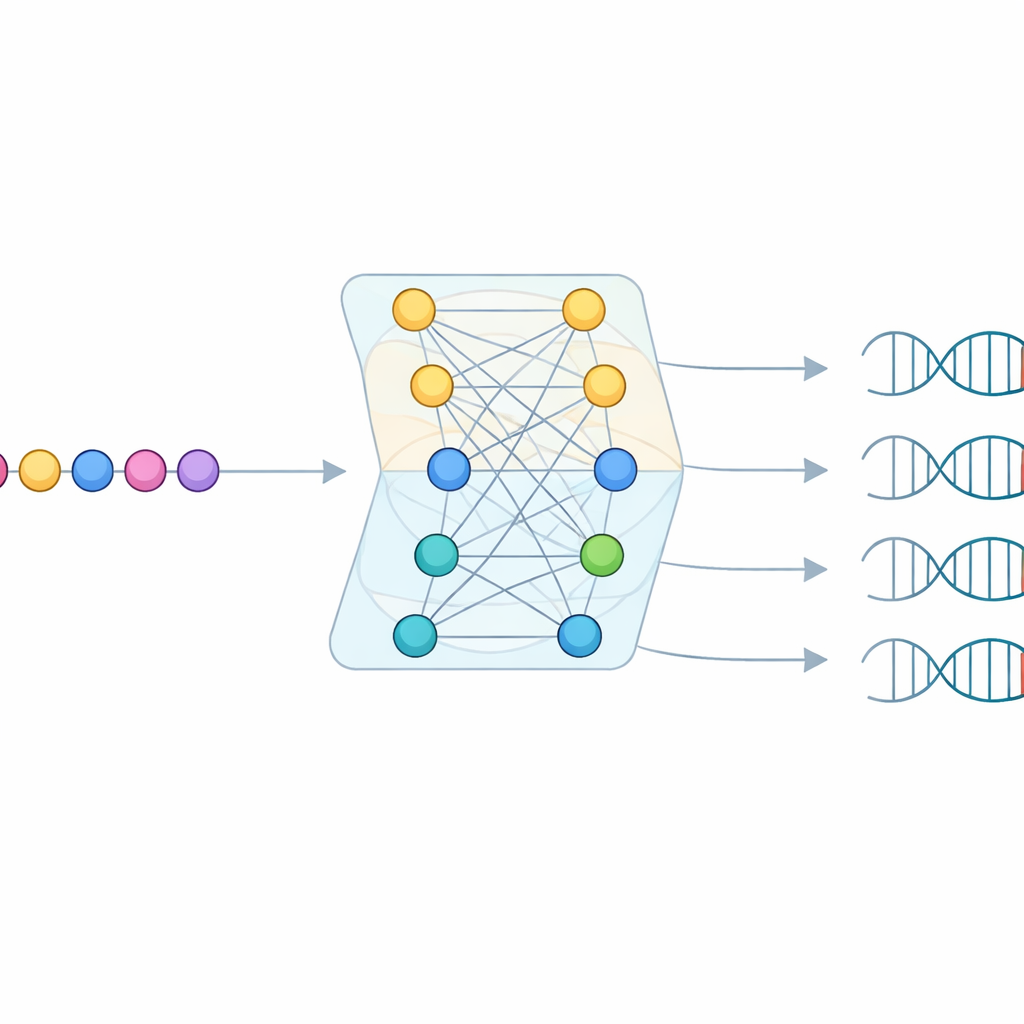
Een belangrijke eigenschap van CICERO is dat het niet alleen een PAM raadt—het schat ook in hoe betrouwbaar elke gok is. Nadat het had geleerd PAM-patronen te voorspellen, trainden de onderzoekers een tweede, lichtgewicht netwerk dat dezelfde Cas9-sequentie gebruikt en leert te voorspellen hoe nauwkeurig de PAM-voorspelling zal zijn. Hogere betrouwbaarheidscores correleerden sterk met hogere werkelijke nauwkeurigheid. Met behulp van dit betrouwbaarheidsfilter breidde het team PAM-annotaties uit naar meer dan 50.000 extra Cas9-eiwitten, waarvan meer dan 17.000 voorspellingen als hoog betrouwbaar werden geclassificeerd. Dit vergroot de keuze aan Cas9-varianten met redelijk goed begrepen targetregels aanzienlijk.
Wat dit betekent voor de behandeling van genetische ziekten
Om te laten zien waarom deze nieuwe bronnen ertoe doen, onderzochten de auteurs tienduizenden ziektegebonden één-lettermutaties in de ClinVar-database die in principe gecorrigeerd zouden kunnen worden met base-editors—instrumenten die één DNA-letter veranderen zonder beide strengen te knippen. Ze vonden dat het standaard Cas9-enzym slechts ongeveer de helft van zulke locaties kan bereiken vanwege zijn strikte PAM-eisen. Wanneer ze Cas9-relaties uit CRISPR-PAMdb en hoogbetrouwbare CICERO-voorspellingen toelieten die een bredere maar nog steeds specifieke set nabije sequenties herkennen, werden vrijwel al deze mutaties theoretisch bereikbaar zonder de targeting zo ver te versoepelen dat precisie verloren zou gaan.
Een grotere gereedschapskist voor precieze DNA-chirurgie
In eenvoudige termen bouwt dit werk twee dingen: een gigantische, openbare kaart die duizenden natuurlijke Cas9-eiwitten verbindt met de korte DNA-patronen die ze prefereren, en een AI-gids die die voorkeuren voor veel meer enzymen alleen op basis van hun sequenties kan voorspellen. Samen veranderen ze de microbiale wereld in een rijke onderdelenbibliotheek voor toekomstige genbewerkers. Naarmate onderzoekers deze Cas9-varianten verfijnen en in het laboratorium testen, kunnen clinici veiligere, veelzijdigere hulpmiddelen krijgen die ziekteveroorzakende mutaties bereiken die eerder buiten bereik waren, en zo werkelijk precieze genoomchirurgie een stap dichterbij brengen.
Bronvermelding: Fang, T., Bogensperger, L., Feer, L. et al. Uncovering Cas9 PAM diversity through metagenomic mining and machine learning. Nat Commun 17, 2510 (2026). https://doi.org/10.1038/s41467-026-69098-5
Trefwoorden: CRISPR-Cas9, PAM-diversiteit, metagenomics, machine learning, genoombewerking