Clear Sky Science · nl
Evaluatie van single-cell ATAC-seq-atlastechnologieën met behulp van sequence-to-function-modellering
Het leesbaar maken van de cellulaire gebruiksaanwijzing
Elke cel in je lichaam leest hetzelfde DNA, maar zenuwcellen, spiercellen en immuuncellen gedragen zich heel verschillend. Dit artikel pakt een centraal raadsel achter die diversiteit aan: hoe korte DNA-streken, enhancers genoemd, als schakelaars fungeren om genen in specifieke celtypen aan of uit te zetten. De auteurs laten zien dat nieuwe, goedkopere labtechnologieën de enorme datasets kunnen leveren die nodig zijn om moderne deep‑learningmodellen te trainen die DNA-sequenties lezen en voorspellen welke enhancers in welke cellen actief zijn, waardoor we dichter bij het echt ontcijferen van de regulatorische “grammatica” van het genoom komen.
Kaarten maken van open DNA in individuele cellen
Enhancers bevinden zich meestal in DNA-streken die meer open en toegankelijk zijn, waardoor regulerende eiwitten er makkelijker aan kunnen binden. Een techniek genaamd single‑cell ATAC‑seq meet welke delen van het genoom open zijn in duizenden tot honderdduizenden individuele cellen tegelijk, en creëert zo een “atlas” van toegankelijk DNA over veel celtypen. Deze atlasgegevens zijn ideale input voor deep‑learningmodellen die ruwe DNA-sequentie als invoer gebruiken en leren te voorspellen hoe sterk elk klein regio als enhancer fungeert in elk celtype. Tot nu toe berustten de meeste van zulke atlasprojecten echter op dure commerciële instrumenten, waardoor de vraag rijst of goedkope, open‑source methoden gelijkwaardige trainingsdata voor deze modellen kunnen leveren.
Een open‑source alternatief voor commerciële platforms
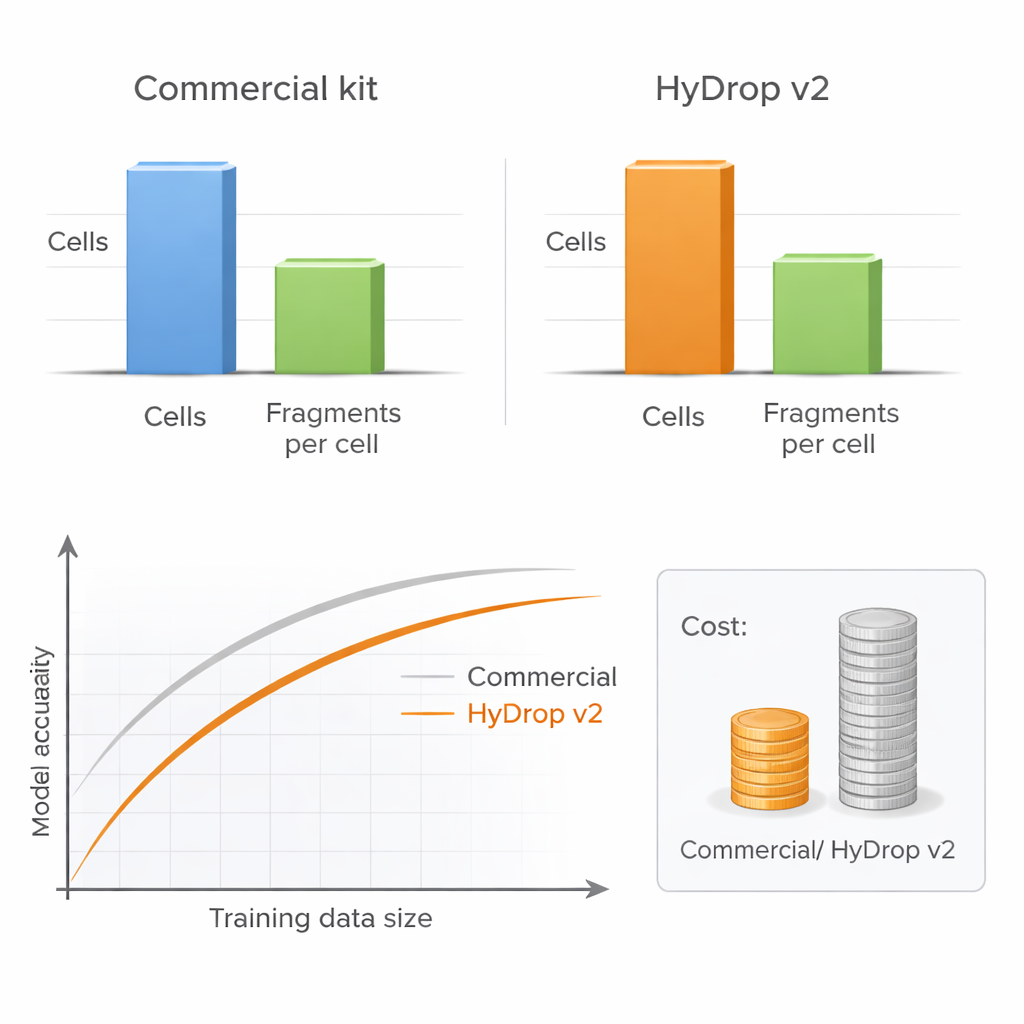
De auteurs introduceren HyDrop v2, een verbeterde druppelgebaseerde methode voor single‑cell ATAC‑seq die aangepaste hydrogelparels gebruikt om individuele cellen te barcoderen. Ze benchmarken HyDrop v2 tegenover een veelgebruikt commercieel kit door grote atlassen te bouwen uit twee heel verschillende systemen: de volwassen muismotorische cortex en laat‑stadium fruitvliegembryo’s. HyDrop v2 genereert vergelijkbare datakwaliteit—het recupereert dezelfde belangrijkste celtypen en zeer vergelijkbare sets van toegankelijke DNA-regio’s—terwijl de kosten per muizenhersenmonster ongeveer veertien keer lager liggen. Belangrijk is dat data uit HyDrop v2-experimenten soepel integreren met commerciële data, zodat onderzoekers platforms kunnen mixen en matchen bij het samenstellen van zeer grote atlassen.
Deep‑learningmodellen trainen om enhancerlogica te lezen
Om te testen of goedkopere data volstaan voor geavanceerde modellering, trainen de onderzoekers sequence‑to‑function deep‑learningmodellen op ofwel commerciële of HyDrop v2‑atlassen. Deze modellen leren rechtstreeks van DNA‑sequentie te voorspellen hoe toegankelijk elke regio is in elk celtype, en kunnen korte sequentiepatronen aanwijzen die waarschijnlijk overeenkomen met bindingsplaatsen voor specifieke regulerende eiwitten. In de muiscortex komen modellen die op HyDrop v2‑data zijn getraind overeen met modellen op commerciële data in algehele nauwkeurigheid en in hun vermogen om bekende enhancer‑“schakelaars” te vinden die eerder in levende dieren zijn gevalideerd. In het vliegembryo ondersteunen beide platforms modellen die kunnen inzoomen op 2.000‑base‑paarregio’s en de kern van ongeveer 500 baseparen aanwijzen die daadwerkelijk weefsel‑specifieke enhanceractiviteit aandrijven, zoals regio’s die neuroblast‑ of spiergenen reguleren.
Meer cellen kunnen dieper sequencen verslaan
Een belangrijke praktische vraag voor elk laboratorium is of ze elke cel zeer diep moeten sequencen of liever meer cellen op lagere diepte moeten profileren. Door systematisch het aantal cellen en het aantal DNA‑fragmenten per cel te variëren, laten de auteurs zien dat de modelprestatie nauwelijks lijdt wanneer de sequencedybte tot een gematigd niveau wordt verlaagd, zolang er maar voldoende cellen worden opgenomen. Daarentegen schaadt het verminderen van het aantal cellen duidelijk de modelnauwkeurigheid, vooral bij het beoordelen van prestaties over veel celtypen tegelijk. Omdat HyDrop v2 per cel veel goedkoper is, kunnen onderzoekers gemakkelijk tienduizenden extra cellen toevoegen en daarmee de prestaties van op commerciële data gebaseerde modellen terugwinnen of zelfs overtreffen tegen een fractie van de kosten.
Proteïne‑voetafdrukken op DNA zichtbaar maken
De studie onderzoekt ook of verschillende labplatforms subtiele vertekeningen inbrengen in hoe het ATAC‑seq‑enzym DNA knipt, wat modellen die proberen te achterhalen waar eiwitten op het genoom zitten zou kunnen misleiden. Met een afzonderlijk neuronaal‑netwerktool dat enzymvoorkeuren corrigeert, tonen de auteurs aan dat HyDrop v2 en commerciële kits vrijwel identieke patronen van enzymactiviteit produceren in zowel muis‑ als vliegcelllen. Na correctie onthullen beide datasets fijnmazige “voetafdrukken” waar regulerende eiwitten en nucleosomen het DNA lijken te beschermen tegen knippen, en deze voetafdrukken komen overeen met de sequentiepatronen die door de sequence‑to‑function‑modellen worden benadrukt. Deze overeenkomst suggereert dat open‑source en commerciële platforms even geschikt zijn voor gedetailleerde studies naar hoe eiwitten met DNA interageren.
Waarom dit belangrijk is voor het ontcijferen van het genoom
Voor niet‑specialisten is de kernboodschap dat we nu zeer grote, betaalbare kaarten kunnen bouwen van hoe DNA in individuele cellen wordt gebruikt, en krachtige deep‑learningmodellen op die kaarten kunnen trainen zonder uitsluitend te leunen op dure, propriëtaire hardware. HyDrop v2 levert data die enhancerpredictie, interpretatie van sequentiepatronen en eiwitbindingsvoetafdrukken ondersteunen op een niveau dat vergelijkbaar is met toonaangevende commerciële methoden, mits er genoeg cellen worden geprofileerd. Dit opent de deur naar het samenstellen van organisme‑brede atlassen van regulatorische elementen in gezondheid en ziekte, waardoor inspanningen om de regulatorische instructies van het genoom te lezen en nieuwe, precies gerichte genetische schakelaars voor onderzoek en toekomstige therapieën te ontwerpen, worden versneld.
Bronvermelding: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Trefwoorden: single-cell ATAC-seq, enhancers, deep learning-modellen, genregulatie, open-source genomica