Clear Sky Science · nl
BiG-SCAPE 2.0 en BiG-SLiCE 2.0: schaalbare, accurate en interactieve sequentieclustering van metabole genclusters
Verborgen chemische schatten in microbieel DNA
Veel van de medicijnen en gewasbeschermingsmiddelen waar we op vertrouwen, zijn afkomstig van kleine moleculen die microben maken. Deze organismen verbergen de recepten voor zulke moleculen in stukken DNA die genclusters worden genoemd. Terwijl DNA-sequencing razendsnel vooruitgaat, verdrinken onderzoekers in data en kennen ze nog maar een fractie van wat microben kunnen produceren. Dit artikel introduceert BiG-SCAPE 2.0 en BiG-SLiCE 2.0, twee geüpgradede softwaretools die wetenschappers helpen enorme genomische archieven te doorzoeken om deze verborgen “moleculaire fabrieken” in kaart te brengen, te vergelijken en te organiseren, en zo de volgende generatie antibiotica en landbouwverbindingen dichter bij ontdekking te brengen.
Waarom genclusters belangrijk zijn voor gezondheid en landbouw
Microben gebruiken gespecialiseerde kleine moleculen om te concurreren, te communiceren en zich aan hun omgeving aan te passen. De DNA-blauwdrukken voor het produceren of afbreken van deze moleculen zijn vaak gegroepeerd in metabole genclusters. Daartoe behoren biosynthetische genclusters die complexe natuurlijke producten opbouwen, en catabolische genclusters die microben in staat stellen bepaalde verbindingen of wortelafscheidingen af te breken. Omdat genen in een cluster samen functioneren, is het vinden van zo’n regio in een genoom vergelijkbaar met het ontdekken van een zelfstandige “fabriekslijn” die een aanwijzing kan geven over de structuur en functie van een molecuul. Genoom-miningtools detecteren dergelijke fabrieken al in bacteriën en schimmels, maar de echte uitdaging is het vergelijken van honderden duizenden clusters om te zien hoe ze gerelateerd zijn en welke chemische diversiteit ze bevatten.
Twee motoren om moleculaire fabrieken te ordenen
BiG-SCAPE en BiG-SLiCE zijn oorspronkelijk ontwikkeld om genclusters met vergelijkbare kernkenmerken in “genclusterfamilies” te groeperen. Van elke familie wordt verwacht dat ze hetzelfde of nauw verwante moleculen produceert. BiG-SCAPE bouwt gedetailleerde netwerken van overeenkomsten tussen clusters, terwijl BiG-SLiCE is afgestemd op snelheid en miljoenen clusters aankan door ze om te zetten in eenvoudige numerieke vingerafdrukken en die vingerafdrukken vervolgens te clusteren. Samen vormen ze de ruggengraat van een groeiend ecosysteem van genoom-miningpijplijnen, databases en interactieve viewers die onderzoekers helpen de microbiële chemie op planetaire schaal te verkennen.
Wat is nieuw in BiG-SCAPE 2.0
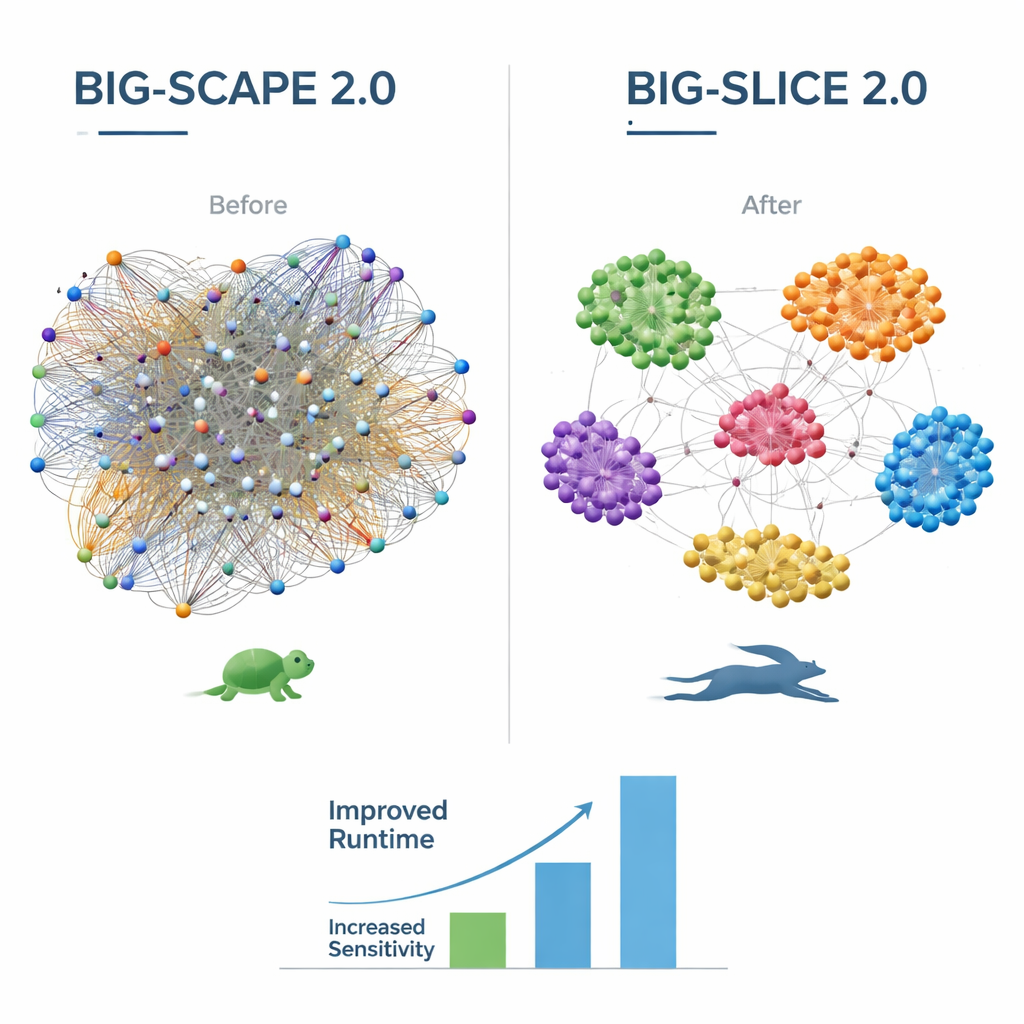
Versie 2.0 van BiG-SCAPE introduceert een reeks verbeteringen gericht op zowel biologie als berekening. Het begrijpt nu het verfijnde “regioconcept” dat door het veelgebruikte antiSMASH-tool wordt gebruikt, waarbij overlappende of hybride genclusters worden opgesplitst in kleinere, betekenisvollere bouwstenen die protoclusters worden genoemd. Nieuwe alignatiemodi en strategieën stellen BiG-SCAPE 2.0 in staat zich te richten op de werkelijk belangrijke kerngenen binnen elk cluster, en beter om te gaan met herschikte genen en vage clustergrenzen. Onder de motorkap is de codebasis volledig herschreven voor snelheid en onderhoudbaarheid, met een gedeelde SQLite-database en een moderne Python-bibliotheek voor profielzoekopdrachten. Als gevolg hiervan kan BiG-SCAPE 2.0 tot acht keer sneller draaien dan zijn voorganger, terwijl het ongeveer de helft van het geheugen gebruikt, en het biedt nu meerdere kant-en-klare workflows voor clusteren, queryen, dedupliceren en benchmarken van genclusters via een verbeterde interactieve webinterface.
Hoe BiG-SLiCE 2.0 de datastroom bijhoudt
BiG-SLiCE 2.0 richt zich op het maken van ultragrote analyses die nauwkeuriger zijn zonder de kenmerkende snelheid te verliezen. Eerdere versies behandelden alle typen genclusters op dezelfde manier, wat onbedoeld sommige families bevoordeelde boven andere. Door over te stappen op een cosine-achtige afstandsmaat en de bibliotheek van biosynthetische proteinsignaturen bij te werken naar de nieuwste standaarden, groepeert BiG-SLiCE 2.0 nu zeer verschillende soorten clusters gelijkmatiger. Codeoptimalisaties en de overstap naar dezelfde snelle profiel-zoekbibliotheek als BiG-SCAPE zorgen voor extra snelheidswinst, en nieuwe opties om alle resultaten als eenvoudige teksttabellen te exporteren maken het makkelijker om BiG-SLiCE in andere analysepijplijnen te integreren. Tests tegen negen datasets met handmatig gecureerde genfamilies tonen aan dat de nauwkeurigheid van BiG-SLiCE 2.0 nu in de buurt komt van die van BiG-SCAPE, vooral voor kortere en moeilijker te detecteren genclusters.
Het onthullen van een enorm, onaangeroerd chemisch universum
De auteurs gebruikten beide tools om 260.630 biosynthetische regio’s uit een publieke database van microbiële genomen te onderzoeken. BiG-SCAPE 2.0 en BiG-SLiCE 2.0 gaven opmerkelijk vergelijkbare schattingen van hoeveel verschillende genclusterfamilies in deze dataset voorkomen, en ondersteunen eerder werk dat aangeeft dat slechts ongeveer 3% van het biosynthetische potentieel gecodeerd in bacteriële genomen tot nu toe is gekarakteriseerd. Met andere woorden: de overgrote meerderheid van microbiëel geproduceerde chemicaliën is onbekend. Door het mogelijk te maken genclusters nauwkeurig te clusteren en te visualiseren over honderden duizenden — en uiteindelijk miljoenen — genomen, bieden BiG-SCAPE 2.0 en BiG-SLiCE 2.0 krachtige lenzen om dit onontgonnen chemische universum te verkennen, en effenen ze het pad voor nieuwe geneesmiddelen, veiligere gewasbeschermingsmiddelen en diepere inzichten in hoe microben ecosystemen en onze eigen gezondheid vormen.
Bronvermelding: Draisma, A., Loureiro, C., Louwen, N.L.L. et al. BiG-SCAPE 2.0 and BiG-SLiCE 2.0: scalable, accurate and interactive sequence clustering of metabolic gene clusters. Nat Commun 17, 2000 (2026). https://doi.org/10.1038/s41467-026-68733-5
Trefwoorden: biosynthetische genclusters, ontdekking van natuurlijke producten, genoom-mining, microbiële metabolieten, computationale clustering