Clear Sky Science · nl
Een alternatieve activatie van EGFR door de R252C-mutatie uit patiëntmateriaal bevordert kankerprogressie
Wanneer cellulaire antennes op hol slaan
Waarom blijven sommige kankers doorgroeien ondanks chemotherapie en geavanceerde immunotherapie? Deze studie volgt een patiënt met tumoren in zowel long als hersenen en herleidt de ziekte tot een kleine wijziging in een belangrijk celoppervlakte-"antenne" genaamd EGFR. Door te onderzoeken hoe die ene mutatie groeisignalen herbedradert, verklaren de onderzoekers niet alleen de agressieve aard van de kanker bij deze patiënt, maar laten ze ook zien hoe een bestaand middel, afatinib, deze activiteit kan temperen.
Een zeldzame mutatie met grote gevolgen
EGFR is een receptor die door het celmembraan heen loopt en cellen helpt te reageren op groeisignalen. Veel long- en hersentumoren dragen mutaties in EGFR, maar de meeste bekende veranderingen zitten binnenin de cel, in het deel dat als een enzymmatige schakelaar fungeert. Hier ontdekten de onderzoekers een ongebruikelijke wijziging aan de buitenkant van EGFR, in het stuk dat normaal groeifactoren bindt. Bij een patiënt met zowel longkanker als glioma vonden ze dat één aminozuur op positie 252 was verwisseld van arginine naar cysteïne — aangeduid als EGFR R252C. Door kanker-databases te doorzoeken, zagen ze deze mutatie bij een klein deel van gliomapatiënten en vrijwel nooit in longtumoren, wat erop wijst dat hij zeldzaam maar reëel is. Met genbewerkingstools recreëerden de auteurs deze exacte mutatie in verschillende menselijke hersen- en longkankercellijnen om te testen wat hij teweegbrengt.
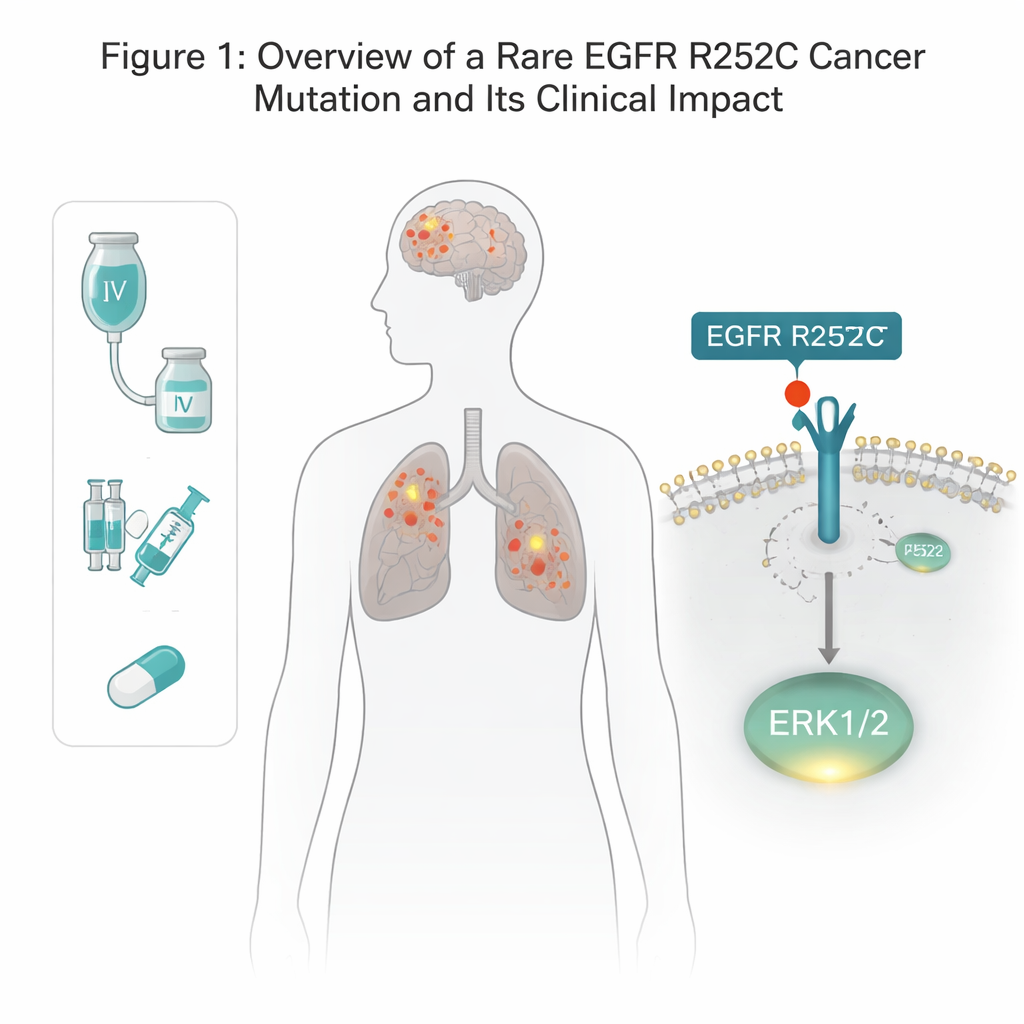
Een nieuwe snelweg naar groeisignalen
Normaal gesproken moet EGFR dimeriseren met een tweede kopie en vervolgens zijn binnenste staart met fosfaatgroepen voorzien voordat het de downstream groeipaden kan inschakelen. Verrassend genoeg vertoonde de R252C-versie dit gebruikelijke zelf-fosforyleringsgedrag niet. In plaats daarvan schakelden cellen met EGFR R252C één specifieke groeiregulator, ERK1/2, veel sterker in dan normaal, terwijl andere klassieke EGFR-routes — zoals AKT en STAT3 — grotendeels onaangetast bleven. Het blokkeren van ERK1/2 met een gerichte remmer maakte het extra groeivoordeel van R252C-cellen ongedaan, waarmee bewezen werd dat ERK1/2 de belangrijkste motor is achter het tumordrijvende vermogen van deze mutant.
De receptor in een altijd-aan houding vergrendelen
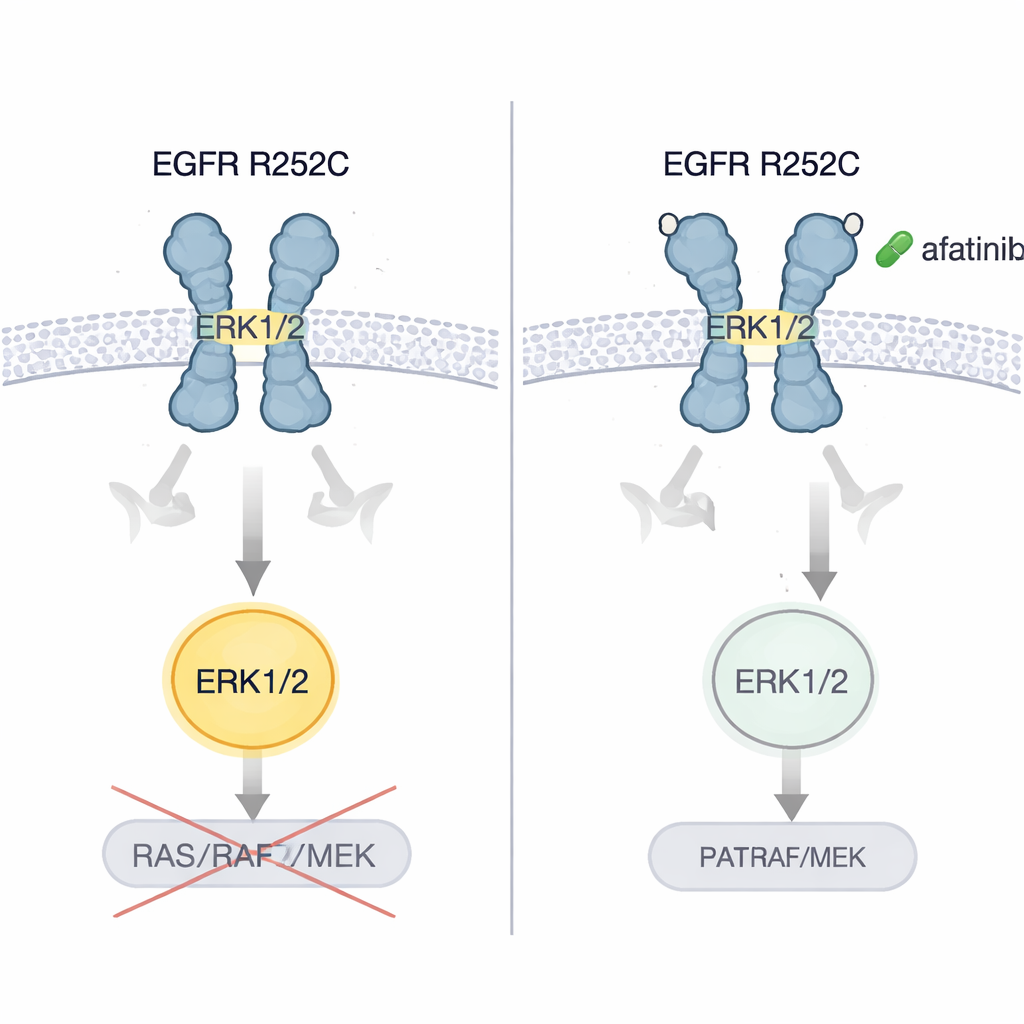
Om te begrijpen hoe een externe verandering zo’n selectieve overactivatie kan veroorzaken, combineerden de onderzoekers biochemische assays met computersimulaties. De R252C-uitwisseling introduceert een nieuwe cysteïne in het buitenste deel van EGFR. Twee van zulke mutanten kunnen een disulfidebrug vormen — een soort moleculaire nietje — tussen hun C252-residuen, waarmee ze stevig aan elkaar worden vastgezet tot een stabiel paar. Structurele modellering toonde dat deze brug de buitenkant van de receptor dwingt in een "V-vormige", verspringende uitlijning die sterk lijkt op de actieve, ligandgebonden toestand, zelfs zonder groeifactor. Deze uitlijning zet zich voort door de membraanspannende en net-binnengelegen segmenten en draait de interne enzymdomeinen in een ongewone configuratie: de actieve zones wijzen naar binnen maar liggen te ver uit elkaar om elkaar efficiënt te fosforyleren. In plaats daarvan creëert deze conformatie een sterke aanhechtingsvlakte voor ERK1/2, waardoor EGFR R252C ERK1/2 rechtstreeks kan fosforyleren en de gebruikelijke RAS–RAF–MEK-relay kan omzeilen.
Van muismodellen tot één enkele patiënt
De auteurs toonden aan dat hersen- en longkankercellen met EGFR R252C sneller groeiden in kweek en grotere, agressievere tumoren vormden wanneer ze in muizen werden geïnmplanteerd, vergeleken met cellen met normale EGFR. Vervolgens testten ze verschillende generaties EGFR-remmers. Alleen afatinib, een remmer van de tweede generatie, schakelde consequent ERK1/2-activatie uit en verminderde sterk de tumorcelgroei. In muismodellen van door R252C aangedreven hersen- en longtumoren remde afatinib de tumoruitbreiding en verlengde het de overleving. Cruciaal: toen de oorspronkelijke patiënt — wiens ziekte was verergerd ondanks chemotherapie, een vaatgerichte behandeling en immunotherapie — werd overgezet op afatinib, lieten scans van zowel long als hersenen duidelijke krimp van de tumorlast zien en genoot de patiënt meerdere jaren zonder progressie.
Wat dit betekent voor patiënten
Dit werk onthult een eerder niet-erkende manier waarop een kanker-veroorzakende EGFR-mutatie kan werken: door twee receptoren buiten de cel aan elkaar te nieten, ze in een actieve pose te draaien die ERK1/2 direct inschakelt in plaats van de gangbare signaalroute te volgen. Voor niet-specialisten is de kernboodschap dat niet alle mutaties in hetzelfde gen zich hetzelfde gedragen, en dat sommige zeldzame veranderingen het meeste baat kunnen hebben bij specifieke bestaande geneesmiddelen. EGFR R252C lijkt tumoren te creëren die bijzonder kwetsbaar zijn voor afatinib. Hoewel deze conclusie momenteel steunt op één gedetailleerd patiëntgeval plus uitgebreid laboratoriumonderzoek, wijst het op meer gepersonaliseerde toetsing van EGFR-mutaties in het buitendomein en suggereert het dat zorgvuldig gekozen gerichte therapieën nieuwe hoop kunnen bieden voor geselecteerde patiënten met moeilijk te behandelen hersen- en longtumoren.
Bronvermelding: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Trefwoorden: EGFR-mutatie, glioma, longkanker, ERK-signaaltransductie, afatinib