Clear Sky Science · nl
Verbeteren van polygeen score-voorspellen voor ondervertegenwoordigde groepen door transfer learning
Waarom uw DNA-risicoscore misschien niet voor u werkt
Genetische "risicoscores" worden steeds vaker gebruikt om iemands kans in te schatten op veelvoorkomende aandoeningen zoals diabetes, hartziekten of hoge bloeddruk. Maar de meeste van deze scores zijn opgebouwd met DNA-gegevens van mensen van Europese afkomst. Daardoor presteren ze vaak slecht voor mensen uit andere achtergronden, wat vragen oproept over eerlijkheid en bruikbaarheid in de klinische praktijk. Deze studie stelt een eenvoudige vraag: kunnen we wat we geleerd hebben van grote Europese datasets hergebruiken om betere, eerlijkere genetische scores voor ondervertegenwoordigde groepen te bouwen — zonder iemands ruwe data te delen?
Van miljoenen DNA-markers naar één risicoscore
Een polygene score is als een rapportcijfer dat de kleine effecten van veel genetische markers over het genoom optelt. Elke marker krijgt een gewicht dat aangeeft hoe sterk hij geassocieerd is met een eigenschap, gebaseerd op grote genetische studies. Wanneer die studies vooral Europeanen omvatten, werkt de resulterende score meestal het beste bij Europeanen. Verschillen in genetische achtergrond — hoe vaak bepaalde DNA-varianten voorkomen en hoe ze samen geërfd worden — betekenen dat dieselde gewichten vaak mislukken bij Afro-Amerikaanse, Hispanische en andere populaties. Het verzamelen van even grote datasets voor elke groep is duur en traag, dus de auteurs kozen voor een machine-learningstrategie genaamd transfer learning: in plaats van bij nul te beginnen voor elke populatie, verfijnen ze een bestaand model dat elders getraind is.
Hoe kennis te lenen zonder ruwe data te delen
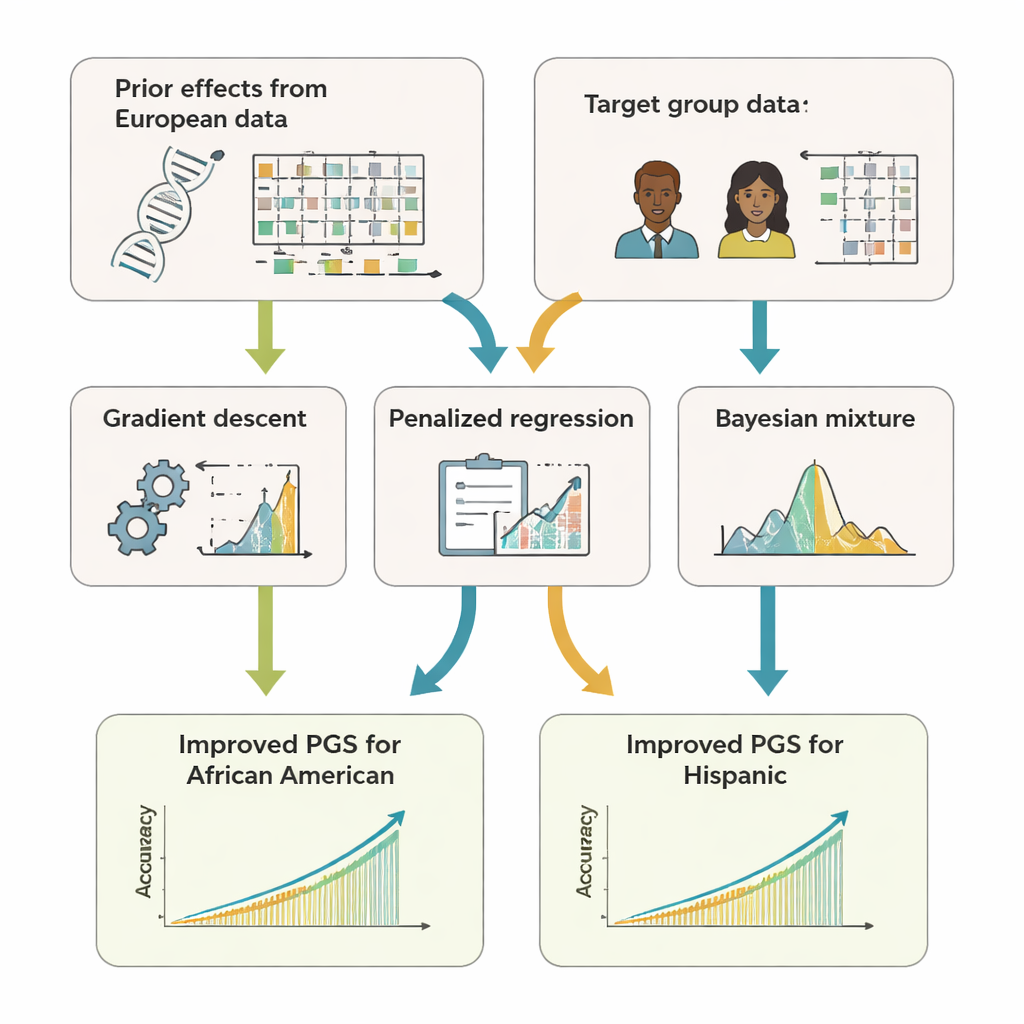
Het team ontwikkelde GPTL, een open-source R-softwarepakket dat drie transfer-learningbenaderingen voor genetische scores implementeert. Alle drie beginnen met bestaande schattingen van DNA-effecten verkregen uit een grote dataset van Europese afkomst en passen die schattingen vervolgens voorzichtig aan met gegevens uit een doeldgroep, zoals Afro-Amerikanen of Hispanics. Een methode past de Europese gewichten stap voor stap aan met gradient descent en stopt vroeg, voordat die volledig overschreven worden. Een tweede methode, genaamd gepenaliseerde regressie, trekt de nieuwe schattingen actief naar de oorspronkelijke waarden tenzij de doeldgegevens sterk bewijs voor het tegendeel leveren. De derde, een Bayesiaans mengmodel, laat elke DNA-marker kiezen uit meerdere informatietoevoeren — zoals meerdere ancestrygroepen of zelfs een "geen-effect" optie — en combineert ze op basis van hoe goed ze de doeldgegevens verklaren.
De methoden op de proef stellen
Om te beoordelen hoe goed deze benaderingen werken, gebruikten de auteurs zowel computersimulaties als echte gegevens van honderden duizenden vrijwilligers in de UK Biobank en het Amerikaanse All of Us-onderzoeksprogramma. Ze richtten zich op Afro-Amerikaanse en Hispanische deelnemers als doeldgroepen en gebruikten gegevens van Europese afkomst als de belangrijkste bron van priorinformatie. Over 11 eigenschappen — waaronder lengte, body mass index, bloedlipiden, bloeddruk en nierwaarden — voorspelden de transfer-learningscores consequent beter dan scores die alleen binnen de doeldgroep waren gebouwd of simpelweg hergebruikt werden van Europeanen. Vaak evenaarden of overtroffen ze zelfs de nauwkeurigheid van complexere "multi-ancestry" methoden die het combineren van ruwe data uit meerdere populaties vereisen. Cruciaal is dat GPTL’s methoden alleen samenvattende statistieken nodig hebben — geaggregeerde getallen over genetische effecten — zodat instellingen kunnen samenwerken zonder individuele genetische gegevens bloot te geven.
Wanneer meer DNA niet altijd beter is
De onderzoekers onderzochten ook hoe je het beste kunt kiezen welke genetische markers op te nemen. In tegenstelling tot de gangbare opvatting dat het gebruik van alle beschikbare markers altijd helpt, vonden ze dat voor Afro-Amerikaanse en vooral Hispanische groepen het opnemen van miljoenen zeer zwakke signalen de prestaties juist kon schaden, vooral bij sterk vereenvoudigde representaties van genetische correlaties. Focussen op beter onderbouwde markers en het gebruiken van rijkere informatie over hoe varianten samen geërfd worden, leverde vaak nauwkeuriger scores op. De studie toonde ook aan dat het toevoegen van priorinformatie uit meerdere ancestrygroepen en het zorgvuldig modelleren van verschillen tussen populaties de voorspellingen verder verbeterde.
Wat dit betekent voor eerlijkere genetische risicovoorspelling
Voor niet-Europese populaties kunnen de kant-en-klare genetische risicoscores van vandaag sterk onderpresteren, wat gezondheidsverschillen kan vergroten. Dit werk laat zien dat transfer learning — het slim verfijnen van bestaande op Europese data gebaseerde scores met bescheiden datasets uit ondervertegenwoordigde groepen — veel van die kloof kan dichten. In de praktijk betekent dit dat zorgsystemen en onderzoekers meer accurate en rechtvaardige genetische hulpmiddelen kunnen bouwen zonder ruwe data tussen instellingen of ancestrieën te poolen, wat privacyzorgen vermindert. Hoewel geen enkele methode voor elk kenmerk en elke populatie het beste zal zijn, laat de GPTL-toolkit zien dat eerlijkere genetische voorspelling technisch haalbaar is als we vorige modellen niet als vaste producten zien, maar als vertrekpunten die voor iedereen aangepast kunnen worden.
Bronvermelding: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Trefwoorden: polygene risicoscores, transfer learning, genetische voorspelling, gezondheidsverschillen, populatiegenetica