Longkankers die resistent zijn tegen EGFR-remmers vertonen collaterale gevoeligheid voor een covalente, cysteïne-onafhankelijke KEAP1-oligomeriserende moleculaire brug
Gerichte geneesmiddelen hebben de behandeling van bepaalde longkankers veranderd door zich te richten op een foutief groeisignaal genaamd EGFR. Voor de meeste patiënten werkt deze therapie echter na een paar jaar niet meer omdat de tumor resistentie ontwikkelt. Deze studie onthult een verrassende wending: zodra tumoren resistent zijn geworden tegen EGFR-remmers, ontstaat er een nieuwe Achillespees die met een ander soort verbinding kan worden aangevallen. Inzicht in deze verborgen zwakte kan toekomstige behandelstrategieën inspireren die de evolutie van kanker in het nauw drijven in plaats van haar steeds na te jagen.
Een verborgen zwakte onthuld
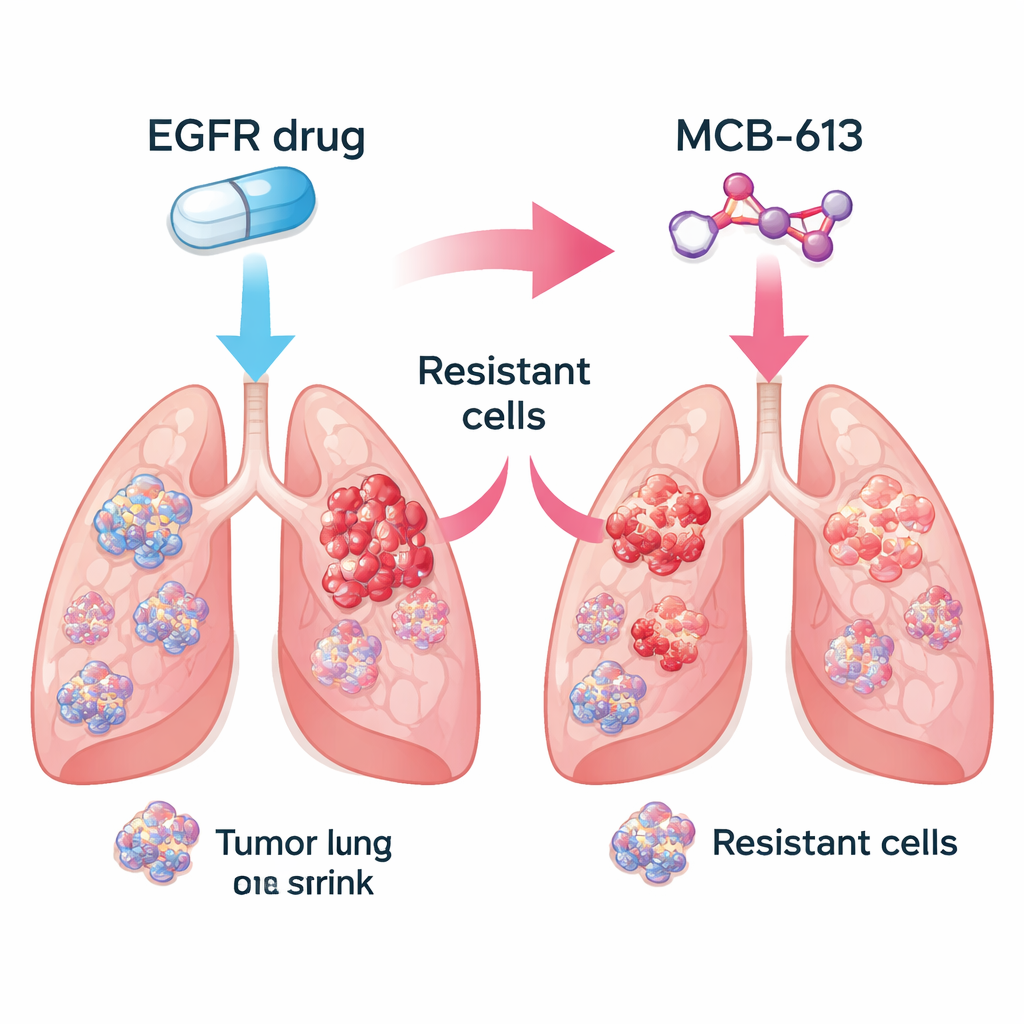
De onderzoekers concentreerden zich op niet-kleincellige longkankers die worden aangedreven door gemuteerd EGFR, een veelvoorkomende vorm van de ziekte. In het laboratorium vergeleken ze medicijngevoelige kankercellen met nauw verwante cellen die resistentie hadden ontwikkeld tegen EGFR-blokkers zoals gefitinib en osimertinib. Vervolgens testten ze een bibliotheek van ongeveer 2.100 kleine moleculen om te zien welke stoffen de resistente cellen effectiever doodden dan de oorspronkelijke, medicijngevoelige cellen. Tussen veel kandidaten stak één verbinding, MCB-613, consequent bovenuit. Resistente cellen die EGFR-remmers konden weerstaan bleken uitzonderlijk kwetsbaar voor MCB-613, zowel in kweek als in muizentumoren.
Gemmengde tumorpopulaties vastzetten Figure 1.
Echte tumoren bestaan uit mengsels van cellen: sommige blijven gevoelig voor het oorspronkelijke middel, terwijl andere via verschillende genetische trucs resistentie verwerven. Het team vroeg zich af of het combineren van een EGFR-remmer met MCB-613 deze diversiteit zou kunnen uitwissen. In een gecontroleerd experiment mengden ze grotendeels medicijngevoelige cellen met een klein aandeel van meerdere resistente types, als een model van een patiëntentumor. Behandeling van deze gemengde populatie met ofwel de EGFR-remmer of MCB-613 alleen liet sommige cellen overleven en groeien. Maar wanneer beide middelen tegelijk werden gebruikt, stortte de gehele populatie in. Dit suggereert dat het combineren van een standaard gerichte therapie met een zorgvuldig gekozen "collaterale gevoeligheids"-medicijn tumoren in een evolutionaire doodlopende straat kan drijven.
Een moleculaire brug die een beschermer breekt
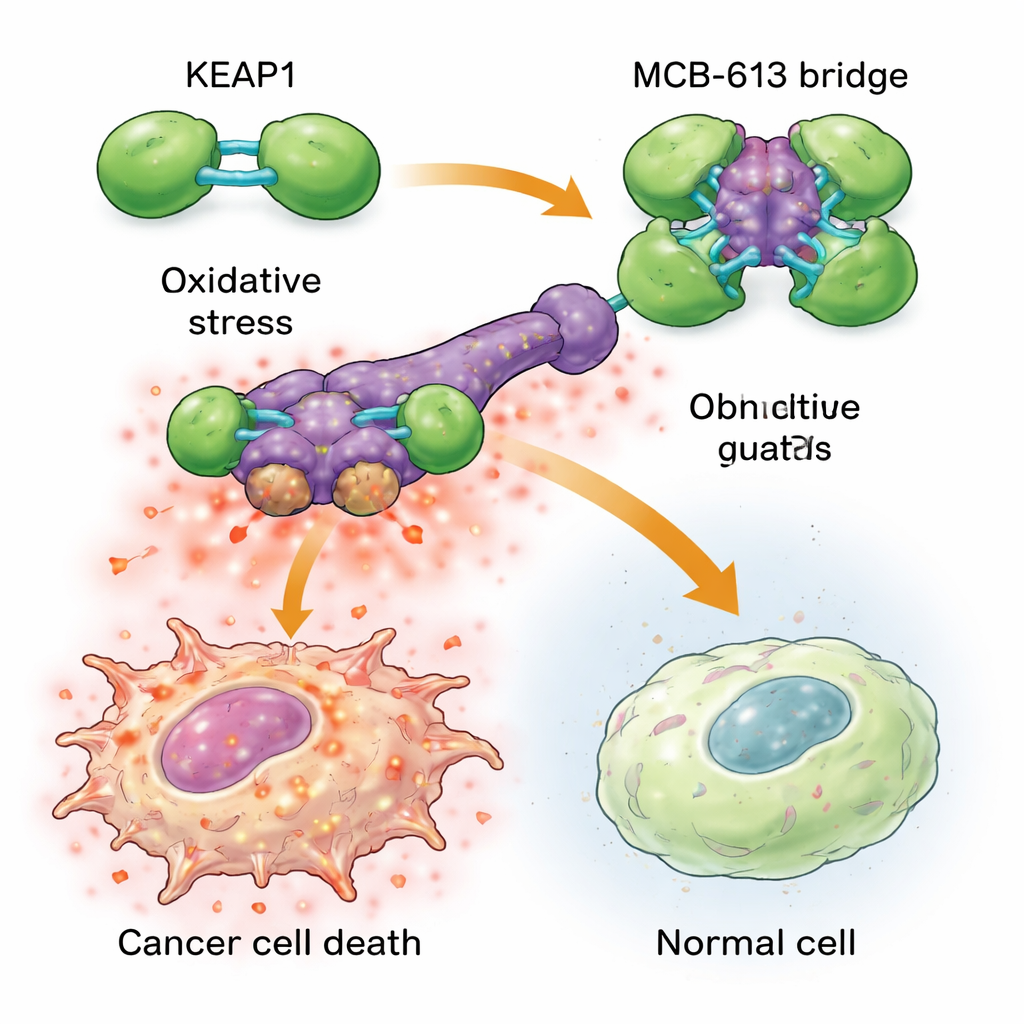
Om te begrijpen waarom MCB-613 resistente cellen zo hard raakt, onderzochten de wetenschappers aan welke eiwitten het bindt. Met behulp van chemische probes en een gerichte CRISPR-genknipscreen identificeerden ze een eiwit genaamd KEAP1 als essentieel voor het effect van MCB-613. KEAP1 fungeert normaal als een cellulair bewaker die stress detecteert en helpt beschermende reacties te reguleren. Het team ontdekte dat MCB-613 zich op een ongebruikelijke manier aan KEAP1 hecht: het gedraagt zich als een starre moleculaire brug die KEAP1-eenheden aan elkaar koppelt tot te grote, abnormale clusters. Dit proces berust niet op de gebruikelijke reactieve zwavelhoudende locaties in KEAP1, maar op een specifiek lysine-aminozuur in het dimerisatiedomein. Wanneer dat lysine gemuteerd was, kon MCB-613 KEAP1 niet langer samenklonteren en waren resistente cellen niet langer overgevoelig voor de verbinding.
Hulpvolle stress omzetten in dodelijke overbelasting Figure 2.
Het samenklonteren van KEAP1 zet een gevaarlijke kettingreactie in gang binnen medicijnresistente kankercellen. Deze cellen leven al onder een hogere achtergrondstress, met verhoogde niveaus van reactieve zuurstofsoorten (schadelijke chemische bijproducten) en een verhoogde activiteit in een beschermend signaalnetwerk dat bekendstaat als de geïntegreerde stressrespons. Wanneer MCB-613 wordt toegevoegd, duwt de verstoring van KEAP1 deze gestreste toestand over de rand: reactieve zuurstof hoopt zich verder op en belangrijke stressregulatoren genaamd ATF4 en CHOP schakelen krachtige doodsprogramma's in. Het blokkeren van deze stressregulatoren, of het chemisch opvangen van reactieve zuurstof, beschermde de cellen grotendeels tegen MCB-613. Interessant genoeg was de klassieke KEAP1-partner NRF2, vaak gezien als de belangrijkste aanjager van antioxidantverdediging, niet verantwoordelijk voor de celdood; het verwijderen van NRF2 maakte de cellen zelfs nog gevoeliger, wat benadrukt dat MCB-613 een andere, niet-canonieke route uitbuit.
Wat dit kan betekenen voor toekomstige behandelingen
Voorlopig is MCB-613 zelf een instrumentele verbinding met chemische nadelen die het ongeschikt maken als geneesmiddel. Maar het onthult een krachtig concept: naarmate longkankers resistentie ontwikkelen tegen EGFR-remmers, kunnen ze vast komen te zitten in een gestreste toestand die selectief kan worden aangepakt door verbindingen die KEAP1 in disfunctionele assemblages dwingen. In principe zouden verfijnde versies van dergelijke "moleculaire bruggen" ontwikkeld kunnen worden die veiliger en preciezer zijn, waardoor oncologen tumoren in een "onmogelijke keuze" kunnen sturen tussen gevoeligheid voor de oorspronkelijke gerichte therapie en gevoeligheid voor de opvolgende stressinducerende stof. Deze evolutionaire valstrategie zou er uiteindelijk toe kunnen bijdragen resistentie bij EGFR-mutante longkanker — en mogelijk ook bij andere moeilijk behandelbare kankers — uit te stellen of te overwinnen.
Bronvermelding: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1