Clear Sky Science · nl
Het gebruik van lineaire referenties uit het pangenoom om ontbrekende autismevarianten te ontdekken
Waarom verborgen DNA-veranderingen belangrijk zijn voor autisme
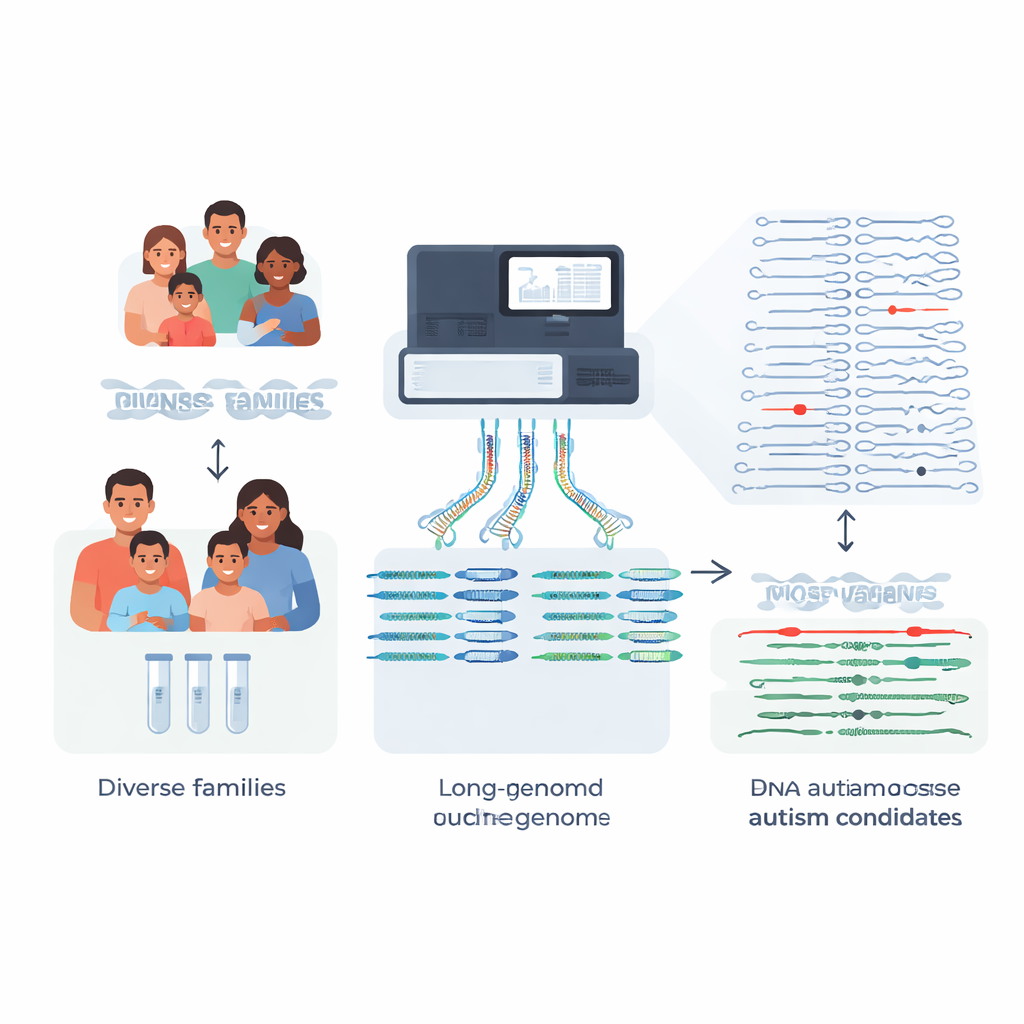
De meeste families die genetisch onderzoek laten doen voor een autistisch kind hopen op duidelijke antwoorden, maar ongeveer vier op de vijf krijgen geen definitieve genetische verklaring. Deze studie pakt een belangrijke reden daarvoor aan: veel invloedrijke DNA-veranderingen zijn te complex voor standaardtests om te detecteren. Door bijna volledige genomen te construeren voor 189 personen uit 51 families met autisme en deze te vergelijken met een nieuwe, rijkere "pangenoom"-referentie, laten de onderzoekers zien hoe geavanceerde sequencing zeldzame, eerder onzichtbare mutaties kan blootleggen die mogelijk enkele gevallen van autisme en verwante aandoeningen kunnen verklaren.
Voorbij standaard genetische tests kijken
Traditionele klinische tests vertrouwen op korte DNA-fragmenten om iemands genoom te doorzoeken. Dat werkt goed voor veel enkelletterveranderingen, maar faalt vaak in repetitieve of structureel complexe gebieden—juist daar waar enkele krachtige ziekteveroorzakende mutaties zich verbergen. Het team richtte zich op families waarbij eerdere short-read genoom-, exoom- of genpaneeltests geen oorzaak hadden gevonden voor autisme of Rett-achtige symptomen. Met long-read sequencing, die veel grotere DNA-stukken leest, bouwden ze hoogwaardige, gefaseerde genoomassemblies voor 189 individuen. Daardoor konden ze van elke persoon beide chromosoomkopieën reconstrueren, één van elke ouder, met zeer weinig ontbrekende stukken.
Structurele varianten: grote veranderingen met grote effecten
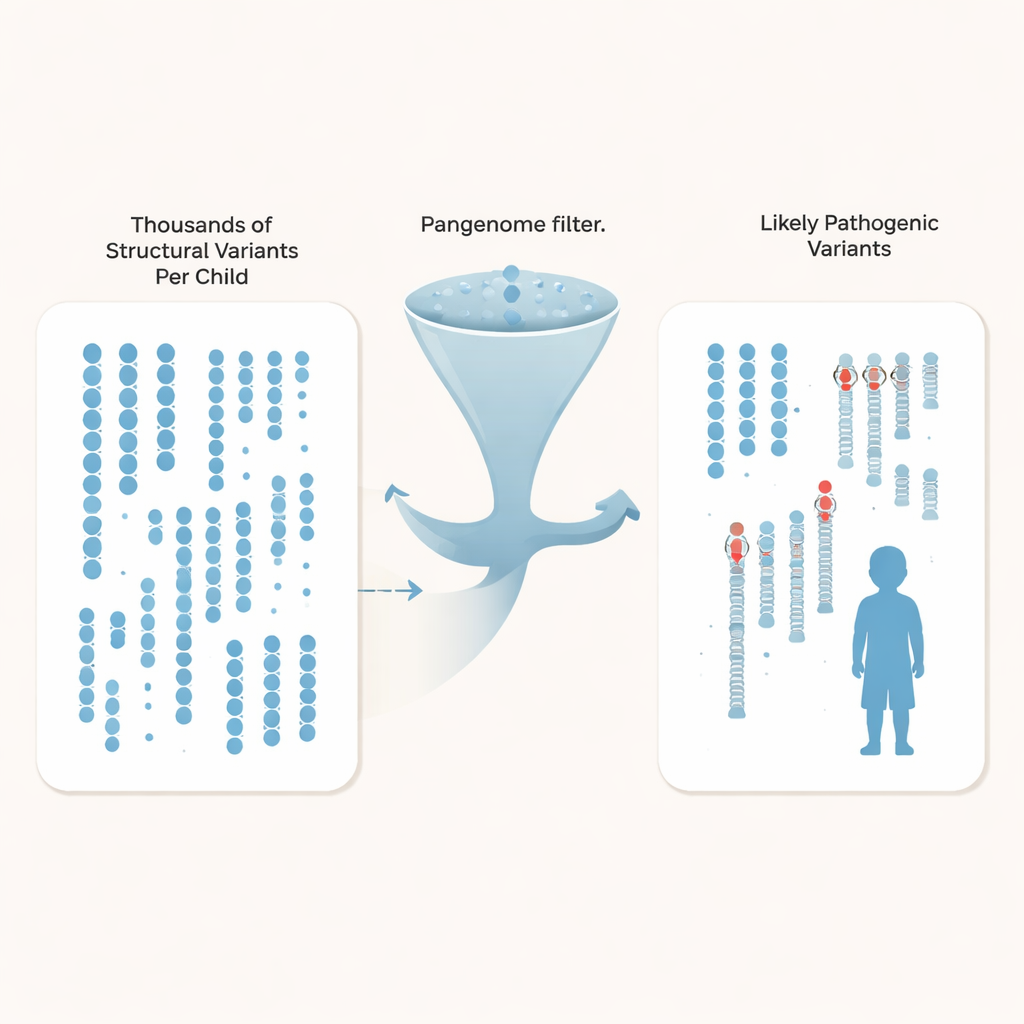
In plaats van alleen enkelletterverschillen te volgen, richtten de onderzoekers zich op structurele varianten—inserties, deleties en herschikkingen die ten minste 50 DNA-letters betreffen en genen of hun regelknoppen kunnen verstoren. Elk kind droeg ongeveer 27.000 van zulke varianten, maar de overgrote meerderheid zijn goedaardige achtergrondverschillen die in de populatie voorkomen. Door hun autismefamilies te vergelijken met honderden diep-geëxamineerde pangenoom-controlegenomen uit diverse ancestries, kon het team meer dan 97% van de gemeenschappelijke structurele varianten voor elk kind filteren, waardoor er ongeveer 600 zeldzame kandidaten per genoom overbleven, en nog weinig—ongeveer 200—bij gebruik van de grootste controleset.
Gemiste mutaties vinden in bekende risicogenen
Met de zoekruimte drastisch verkleind, combineerden de auteurs verschillende bewijslijnen: bekende autisme- en neuro-ontwikkelingsstoornisgenen, regelgebieden actief in de ontwikkelende menselijke cortex, en overervingspatronen binnen elk gezin. Ze ontdekten drie duidelijk pathogene mutaties die eerdere tests hadden gemist. Daartoe behoorde een nieuw stopsignaal in het SYNGAP1-gen, dat belangrijk is voor synapsfunctie, en een deletie die het laatste exon van MECP2 wegsnijdt, een sleutelgen voor het Rett-syndroom, ondanks dat de patiënt meerdere eerdere klinische tests had ondergaan. Ze bevestigden ook een ziekteveroorzakende verandering in TBL1XR1, een gen dat met MECP2 interageert. In totaal belichtten ze negen aanvullende structurele varianten—vaak geërfd en gelegen in regelgebieden nabij hersengerelateerde genen—als sterke kandidaten voor toekomstig functioneel onderzoek.
Wat de studie niet vond—en waarom dat toch belangrijk is
Ondanks deze grondige zoektocht zagen de auteurs geen duidelijke algehele toename van structurele varianten bij autistische kinderen in vergelijking met hun niet-aangedane broers of zussen, althans in deze bescheiden steekproefomvang. Er was echter een aanwijzing voor meer structurele veranderingen op het X-chromosoom bij getroffen meisjes, en de vrijwel complete X- en Y-assemblies stelden hen in staat ongebruikelijke patronen op te merken zoals extreme scheefheid in X-chromosoominactivatie. Deze kenmerken kunnen belangrijke aanwijzingen worden naarmate meer families onderzocht worden. Cruciaal is dat het werk aantoont dat long-read sequencing pathogene varianten kan terugvinden die short-read methoden missen, vooral in lastige delen van het genoom en in controlegebieden die de genactiviteit fijnregelen.
Wat dit betekent voor families en toekomstig onderzoek
Voor families is de directe impact bescheiden maar van betekenis: binnen deze moeilijk-op-te-lossen gevallen kreeg ongeveer 6% een duidelijke genetische diagnose, en bijna één op de vijf kreeg sterke nieuwe kandidaatvarianten om te onderzoeken. Voor het veld is de boodschap groter. Naarmate meer diverse, complete referentiegenomen aan het pangenoom worden toegevoegd en long-read sequencing toegankelijker wordt, zullen clinici gemeenschappelijke structurele veranderingen kunnen uitsluiten en snel kunnen focussen op een kleine set zeldzame, mogelijk schadelijke varianten bij elke patiënt. Die verschuiving zou geleidelijk veel van de huidige “onopgeloste” autismegevallen kunnen veranderen in gevallen waarbij de onderliggende biologie—en mogelijke paden naar ondersteuning en behandeling—veel beter begrepen zijn.
Bronvermelding: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Trefwoorden: autismegenetica, long-read sequencing, structurele varianten, menselijk pangenoom, Rett-syndroom