Clear Sky Science · nl
Best practices en hulpmiddelen in R en Python voor statistische verwerking en visualisatie van lipidomics- en metabolomicsgegevens
Waarom het omzetten van labcijfers naar heldere beelden ertoe doet
Moderne instrumenten kunnen nu duizenden kleine moleculen—lipiden en andere metabolieten—meten in een enkele druppel bloed of weefsel. Deze metingen bevatten aanwijzingen over ziekerisico's, behandelreacties en hoe ons lichaam reageert op voeding of veroudering. Maar de ruwe output is geen kant-en-klaar antwoord: het is een enorme tabel met cijfers die schoongemaakt, geanalyseerd en omgezet moeten worden in begrijpelijke beelden. Dit artikel legt uit hoe onderzoekers twee populaire programmeertalen, R en Python, kunnen gebruiken om dit betrouwbaar, transparant en met publicatieklare grafieken te doen.
Van chemische metingen naar complexe datatabellen
In lipidomics en metabolomics genereren massaspectrometrie en chromatografie grote datasets waarbij elke rij een sample is en elke kolom een molecuul. Deze tabellen gedragen zich zelden als voorbeeldtabellen uit lesboeken. Ze bevatten missende waarden, uitschieters en scheve verdelingen waarbij enkele moleculen extreem hoge waarden vertonen. Concentraties kunnen meerdere grootteordes bestrijken en worden beïnvloed door leeftijd, geslacht, voeding, medicatie, dagelijkse ritmes en technische problemen zoals instrumentdrift of batcheffecten. Internationale expertengroepen hebben richtlijnen uitgebracht om te standaardiseren hoe monsters worden verzameld, verwerkt en gerapporteerd, maar zelfs met goede laboratoriumpraktijken blijft zorgvuldige statistische verwerking essentieel om echte biologische signalen uit deze rumoerige achtergrond te halen.
Het schoonmaken en voorbereiden van de cijfers
Voordat vergelijkingen tussen gezonde en zieke groepen betekenis krijgen, moeten de gegevens worden voorbereid. De review beschrijft hoe missende waarden ontstaan—door willekeurige fouten, beperkingen van het instrument of signaalinterferentie—and legt uit wanneer ze veilig genegeerd kunnen worden, wanneer opnieuw gemeten moet worden en hoe ze zinnig geschat (geïmputeerd) kunnen worden met methoden zoals k-nearest neighbors, random forests of eenvoudige vervanging door een lage waarde. Vervolgens schetsen de auteurs normalisatiestrategieën die ongewenste variatie verminderen, bijvoorbeeld door batcheffecten te corrigeren met kwaliteitscontrolemateriaal of te corrigeren voor verschillen in monsterhoeveelheid. Daarna bespreken ze transformaties zoals logaritmen—die lange rechtse staarten in de data temmen—en schaalmethoden die alle moleculen op gelijke voet zetten zodat sterk variabele verbindingen later analyses niet domineren. 
Statistische tests en visuele verhalen
Als de gegevens correct zijn voorbereid, komt een reeks statistische hulpmiddelen in beeld. Voor individuele moleculen kunnen onderzoekers fold changes berekenen en klassieke tests gebruiken zoals de t-toets of bij niet-parametrische alternatieven (zoals de Mann–Whitney-toets) om te vragen of niveaus tussen groepen verschillen. Voor vergelijkingen met meerdere groepen worden methoden zoals ANOVA of de Kruskal–Wallis-toets geïntroduceerd, vergezeld van post-hocprocedures om te bepalen welke groepen van elkaar verschillen. De kracht van deze tests komt tot leven wanneer de resultaten helder gevisualiseerd worden. Het artikel licht boxplots toe (inclusief verbeterde versies voor scheve data), violinplots en vulkaanplots die effectgrootte en statistische significantie combineren. Voor lipiden worden meer gespecialiseerde visualisaties beschreven, zoals lipidenetwerken die gecoördineerde veranderingen binnen klassen tonen, en fatty acyl chain-plots die patronen in koolstofketenlengte en verzadiging onthullen.
Patronen zien in veel variabelen tegelijk
Aangezien elk monster honderden of duizenden gemeten moleculen kan hebben, zijn multivariate methoden cruciaal. De review legt uit hoe principal component analysis (PCA) deze complexiteit comprimeert tot een paar nieuwe assen die de belangrijkste variatierichtingen vangen, wat snelle controles mogelijk maakt voor groepsscheiding, batcheffecten of analytische stabiliteit. Geavanceerdere niet-lineaire methoden, waaronder t-SNE en UMAP, kunnen subtiele clusters en structuren in hoge-dimensionale ruimte onthullen. Voor situaties waarin het doel is om monsters te classificeren—bijvoorbeeld het onderscheiden van patiënten en controles—beschrijven de auteurs gesuperviseerde benaderingen gebaseerd op Partial Least Squares en de orthogonale uitbreiding daarvan (PLS-DA en OPLS-DA). Deze methoden koppelen moleculaire profielen aan samplelabels, ondersteunen feature-selectie en worden vaak samengevat met scoreplots, loadingplots en receiver operating characteristic-curven.
Praktische toolkits in R en Python
Om beginners te helpen van theorie naar praktijk te gaan, geeft het artikel een overzicht van een breed ecosysteem aan softwarepakketten. In R vereenvoudigen collecties zoals tidyverse en tidymodels data-wrangling en modellering, terwijl ggplot2 en aanvullende pakketten zoals ggpubr, ggstatsplot en tidyplots het makkelijker maken publicatieklare figuren te genereren. Gespecialiseerde libraries behandelen PCA, clustering en PLS-gebaseerde modellen, en Bioconductor-pakketten ondersteunen complexe heatmaps en interactieve visualisaties. In Python biedt pandas tabelverwerking, terwijl matplotlib, seaborn en plotly visualisatie dekken en scikit-learn een breed scala aan multivariate methoden levert. Doorlopend benadrukken de auteurs stapsgewijze voorbeelden in een bijbehorende GitBook, zodat lezers workflows kunnen reproduceren en aanpassen aan hun eigen data. 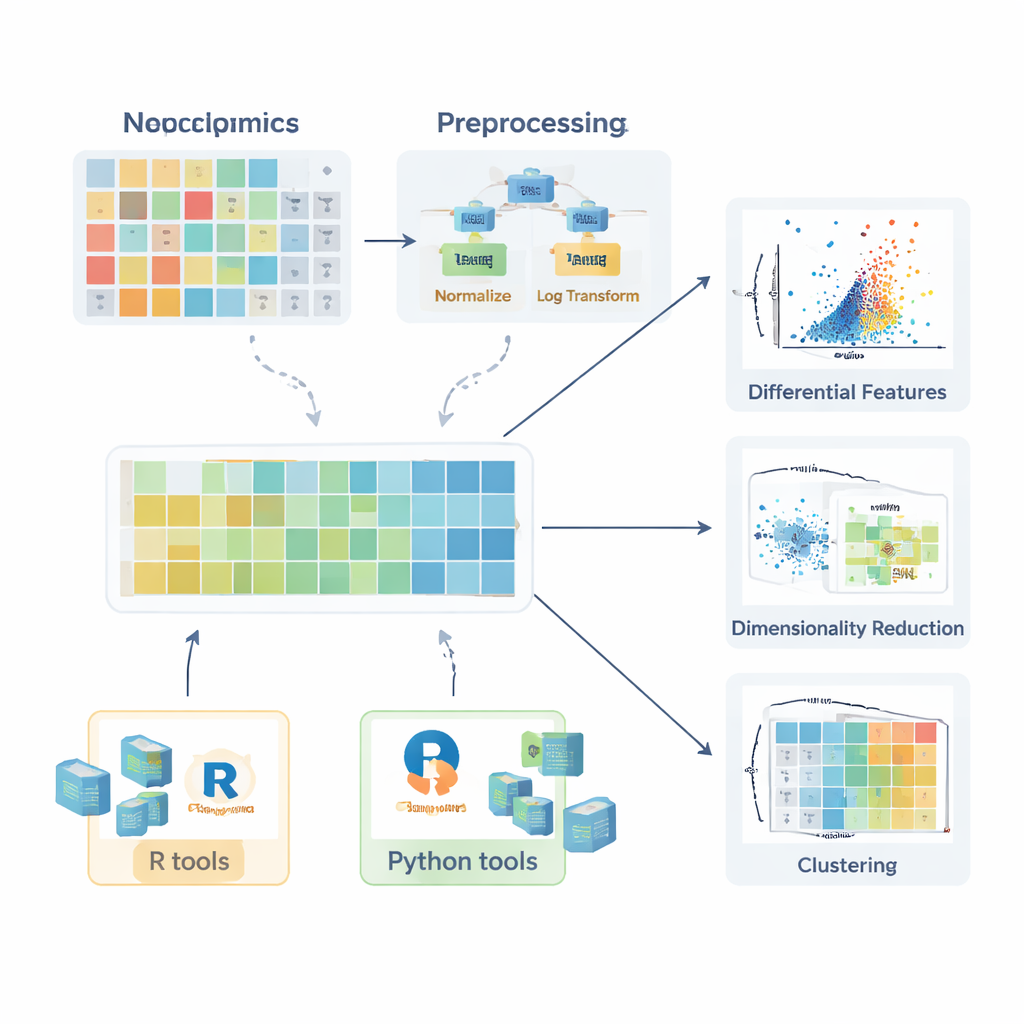
Complexe chemie omzetten in betrouwbare inzichten
Het artikel besluit dat de echte belofte van lipidomics en metabolomics niet alleen in krachtige instrumenten ligt, maar in hoe doordacht hun output wordt verwerkt en gevisualiseerd. Door goede statistische praktijk te volgen, open en goed gedocumenteerde hulpmiddelen in R en Python te gebruiken en te leunen op gedeelde codevoorbeelden, kunnen onderzoekers robuuste en reproduceerbare pipelines bouwen. Dat vergroot de kans dat patronen gevonden in kleine moleculen zich vertalen naar betrouwbare biomarkers, een beter begrip van ziektemechanismen en meer gepersonaliseerde benaderingen van de geneeskunde die uiteindelijk patiënten ten goede komen.
Bronvermelding: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Trefwoorden: lipidomics, metabolomics, datavisualisatie, R-programmering, Python