Clear Sky Science · nl
Ruimtelijke transcriptomica combineren met weefselmorfologie
Het weefsel binnenstebuiten bekijken op twee manieren
Artsen en onderzoekers willen steeds vaker niet alleen weten welke genen in een weefsel actief zijn, maar precies waar ze zijn aangezet. Tegelijkertijd maken ziekenhuismicroscopen al rijke beelden van weefselstructuur die pathologen dagelijks gebruiken. Dit artikel legt uit hoe een nieuw vakgebied probeert deze twee gezichtspunten te verbinden — gedetailleerde kaarten van genactiviteit en gewone microscoopfoto’s — en waarom die combinatie kan leiden tot eerder gediagnosticeerde aandoeningen, betere tumorgradering en dieper inzicht in hoe ziekten zich ontwikkelen en verspreiden.
Van verspreide cellen naar kaarten van genactiviteit
Jarenlang vereisten krachtige “omics”-methoden dat weefsels werden vermalen tot een mengsel van losse cellen, waardoor de informatie over waar elke cel vandaan kwam verloren ging. Ruimtelijke transcriptomica veranderde dat door genactiviteit te meten terwijl de positie van elke cel in het weefsel behouden bleef. Het resultaat is een raster van plekken, elk met een profiel van genexpressie en nauwkeurige coördinaten. Op zichzelf heeft deze ruimtelijke geninformatie al nieuwe patronen van cellulaire diversiteit en ziekte‑architectuur aan het licht gebracht. Maar de meting verandert niet nadat ze is gedaan, en het herhalen van het experiment is duur. Ter vergelijking: weefselbeelden gekleurd met standaardkleuringen, zoals het veelgebruikte hematoxyline en eosine (H&E), zijn goedkoop en overvloedig en bevatten visuele aanwijzingen over celvorm, dichtheid en weefselorganisatie.
Twee manieren om beelden en genen te combineren
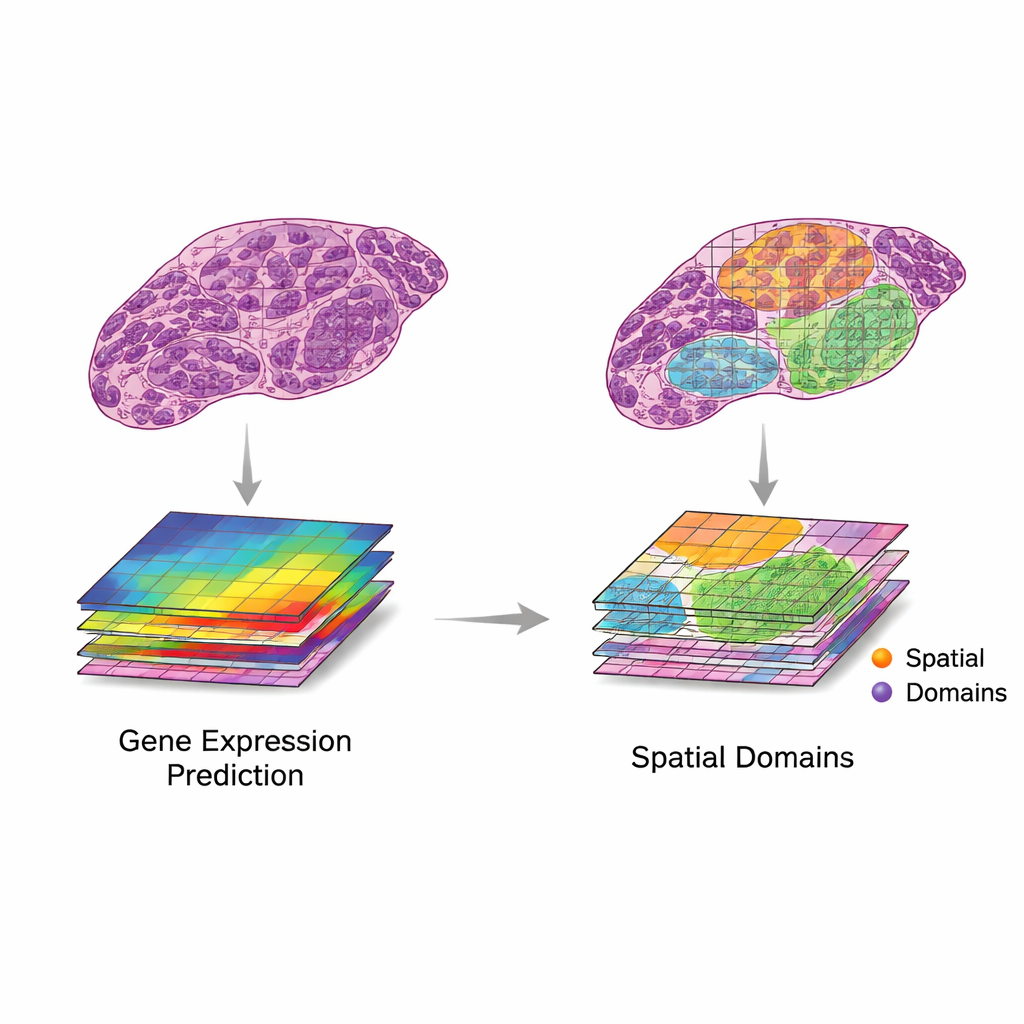
De review stelt een eenvoudig maar krachtig kader voor om deze twee gegevensbronnen samen te gebruiken. Eerst worden beeldpatches gekoppeld aan nabijgelegen plekken met genexpressie. Vervolgens halen computermodellen kenmerken uit beelden — patronen die vorm, textuur en organisatie vangen — en vergelijken die met patronen in genexpressie. De auteurs beschrijven twee wenselijke scenario’s. Bij “vertaling” volgen beeldkenmerken relevante genactiviteit nauwgezet, waardoor modellen kunnen voorspellen welke genen actief zijn op basis van alleen het weefselbeeld. Dit kan worden gebruikt om ontbrekende genmetingen aan te vullen, een hogere resolutie te bereiken dan het oorspronkelijke raster, of genactiviteit af te leiden uit routinematige klinische preparaten zonder extra laboratoriumwerk. Bij “integratie” vangen beeldkenmerken bruikbare informatie die gengegevens missen, zoals trage structurele veranderingen of subtiele weefselorganisatie, wat helpt duidelijkere regio’s of “domeinen” binnen een weefsel te definiëren.
Wanneer extra informatie helpt — en wanneer ze schaadt
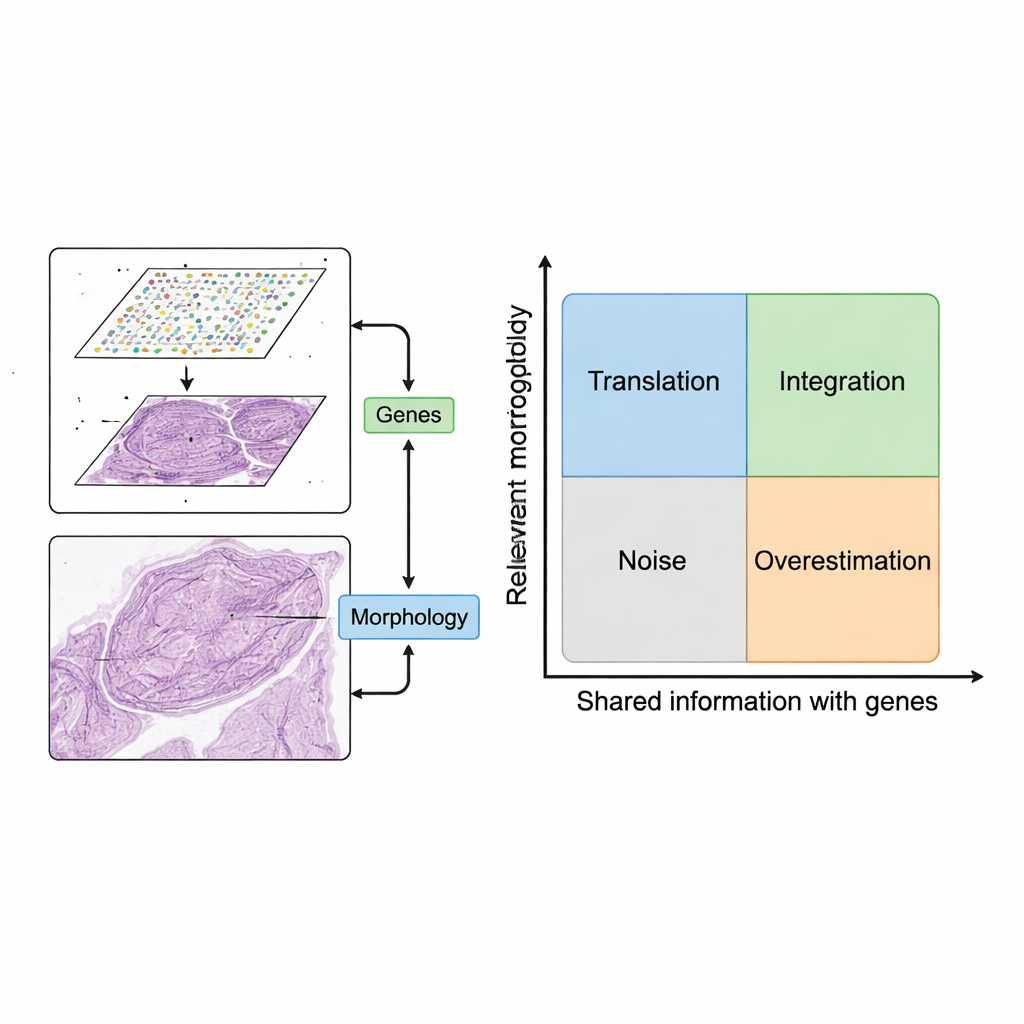
Niet elk beeldkenmerk is de moeite waard om te gebruiken. De auteurs introduceren een conceptuele kaart met twee assen: hoe relevant een beeldkenmerk is voor de biologische vraag, en hoeveel overlap het heeft met geninformatie. Kenmerken die noch relevant noch gerelateerd aan genen zijn, komen neer op ruis, zoals kleuringsartefacten. Kenmerken die genpatronen volgen maar gekoppeld zijn aan onbelangrijke genen (zoals basale housekeepinggenen) kunnen modellen op papier goed laten lijken terwijl ze weinig klinische waarde toevoegen. Door methoden in vier kwadranten te organiseren — vertaling, integratie, ruis en overschatting — maakt het kader duidelijk wanneer het combineren van beelden en genen echt nieuw inzicht oplevert en wanneer het simpelweg herhaalt of verdoezelt wat al bekend is.
Huidige tools, tests en groeipijnen
Een snelgroeiende golf van kunstmatige-intelligentie-methoden probeert nu vertaling en integratie op echte data uit te voeren. Vroege systemen vertrouwden op convolutionele neurale netwerken, terwijl nieuwere systemen transformers, grafneurale netwerken en multi‑schaalmodellen gebruiken die details van kleine celstructuren tot context van hele preparaten kunnen verwerken. Deze methoden zijn gebruikt om genactiviteit uit H&E‑beelden te voorspellen, superresolutiekaarten te genereren en te helpen bij het identificeren van weefselregio’s met verschillend gedrag. Om prestaties te beoordelen, vertrouwen onderzoekers op statistische maatstaven zoals correlatie tussen voorspelde en waargenomen genniveaus, of overeenstemming tussen AI‑gedefinieerde regio’s en labels van ervaren pathologen. Datasets zijn echter nog klein en gevarieerd, en vergelijking tussen studies is moeilijk. Veel gerapporteerde winst kan het gevolg zijn van overfitting, of succes op genen en patronen die in de klinische praktijk weinig betekenen.
Waar dit toe kan leiden
De auteurs concluderen dat het combineren van ruimtelijke genkaarten met weefselbeelden veelbelovend maar nog in een vroeg stadium is. Hedendaagse modellen behalen vaak slechts matige nauwkeurigheid en zijn nog niet klaar voor routinematig medisch gebruik. Toekomstige vooruitgang zal waarschijnlijk komen van betere beeldkenmerken, met name grote “foundation‑modellen” getraind op miljoenen pathologiedia’s, en van een focus op genen en patronen die werkelijk invloed hebben op de patiëntenzorg. Zorgvuldig ontworpen integratie zou op een dag vroege waarschuwingssignalen van ziekte kunnen onthullen door afwijkingen te herkennen tussen hoe het weefsel er nu uitziet en wat de genen voorspellen dat er later zal gebeuren. Kortom, dit werk schetst een routekaart om routinematige microscoopbeelden om te vormen tot rijke, gengeïnformeerde kaarten die artsen helpen ziekte preciezer te begrijpen en te behandelen.
Bronvermelding: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Trefwoorden: ruimtelijke transcriptomica, weefselmorfologie, digitale pathologie, genexpressievoorspelling, beeldvormings-AI