Clear Sky Science · nl
Het activeren van KLF15 in cardiomyocyten: een nieuwe aanpak om pathologische herprogrammering en fibrose te voorkomen via nuclease-deficiënte dCas9VPR
Het herprogrammeren van het falende hart
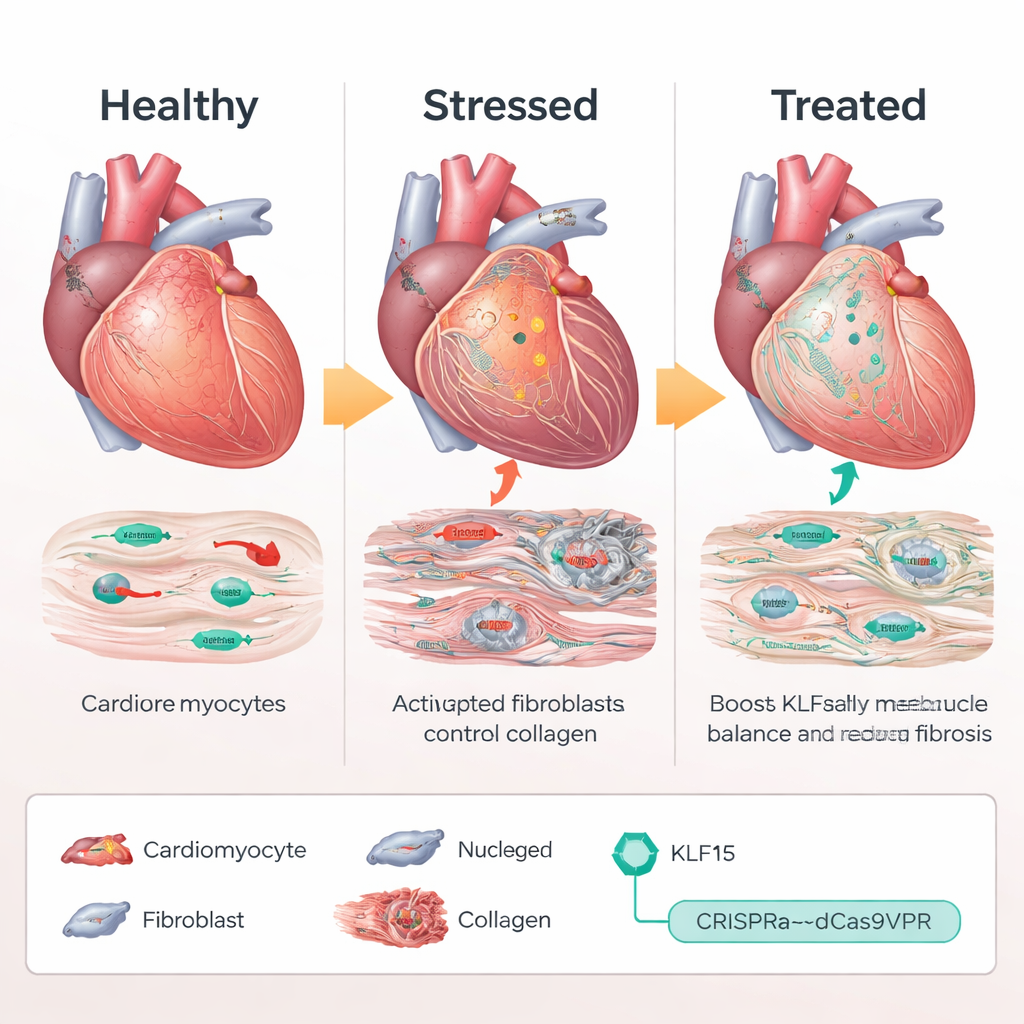
Hartfalen treft miljoenen mensen en ontwikkelt zich vaak langzaam na jaren van hoge bloeddruk of klepziekte. In deze situaties worden hartspiercellen niet alleen groter, ze schakelen ook een "foetaal" genprogramma in en het hart vult zich met littekenweefsel. Deze studie onderzoekt een nieuwe manier om de eigen genregelende mechanismen van het hart weer richting gezondheid te sturen — zonder het DNA te knippen — door voorzichtig een beschermende regelaar genaamd KLF15 in hartspiercellen op te voeren.
Wanneer hartcellen hun identiteit verliezen
In een gezond volwassen hart verbranden cardiomyocyten — hartspiercellen — efficiënt vetten voor energie en behouden ze een stabiel patroon van genactiviteit. Met behulp van single-cell RNA-sequencing bij muizen die chronische drukbelasting ondergingen, brachten de onderzoekers in kaart hoe individuele cardiomyocyten veranderen terwijl het hart van normale functie naar vergroting en uiteindelijk naar falen beweegt. Ze vonden dat een transcriptiefactor genaamd KLF15, die normaal gesproken metabolisme en groei in balans houdt, de sterkste activiteitverandering vertoonde in zieke cellen. Naarmate de stress toenam, daalden de KLF15-niveaus en verzwakte zijn vermogen om foetale en stressgerelateerde genen in toom te houden. Vergelijkbare dalingen van KLF15 werden gezien in menselijke harten van patiënten met gedilateerde en hypertrofische cardiomyopathie, wat aangeeft dat deze verstoring over soorten heen bewaard blijft.
CRISPR als volumeknop, niet als schaar
In plaats van een extra kopie van het KLF15-gen toe te voegen of het DNA te knippen, gebruikte het team een op CRISPR gebaseerde "activatie"-systeem, dCas9VPR, dat bindt in de buurt van het natuurlijke Klf15-gen en de eigen expressie versterkt. In muizen die zo waren geconstrueerd dat deze CRISPR-activator alleen in cardiomyocyten tot expressie kwam, leverden de wetenschappers gids-RNA's af met een adeno-geassocieerd virus (AAV9) om het Klf15-promotergebied te targeten. Bij chronische drukbelasting hielden muizen die Klf15-activerende gidsen ontvingen vrijwel normale Klf15-niveaus. Hun hartspiercellen bleven kleiner, de pompfunctie ging minder achteruit en de overleving verbeterde vergeleken met controledieren. Op moleculair niveau werden stress- en foetale genen tot rust gebracht, terwijl metabolisme- en calciumregulerende genen herstelden, wat aangeeft dat het ongezonde transcriptieprogramma grotendeels werd teruggezet.
Het dempen van littekenvorming via crosstalk tussen cellen
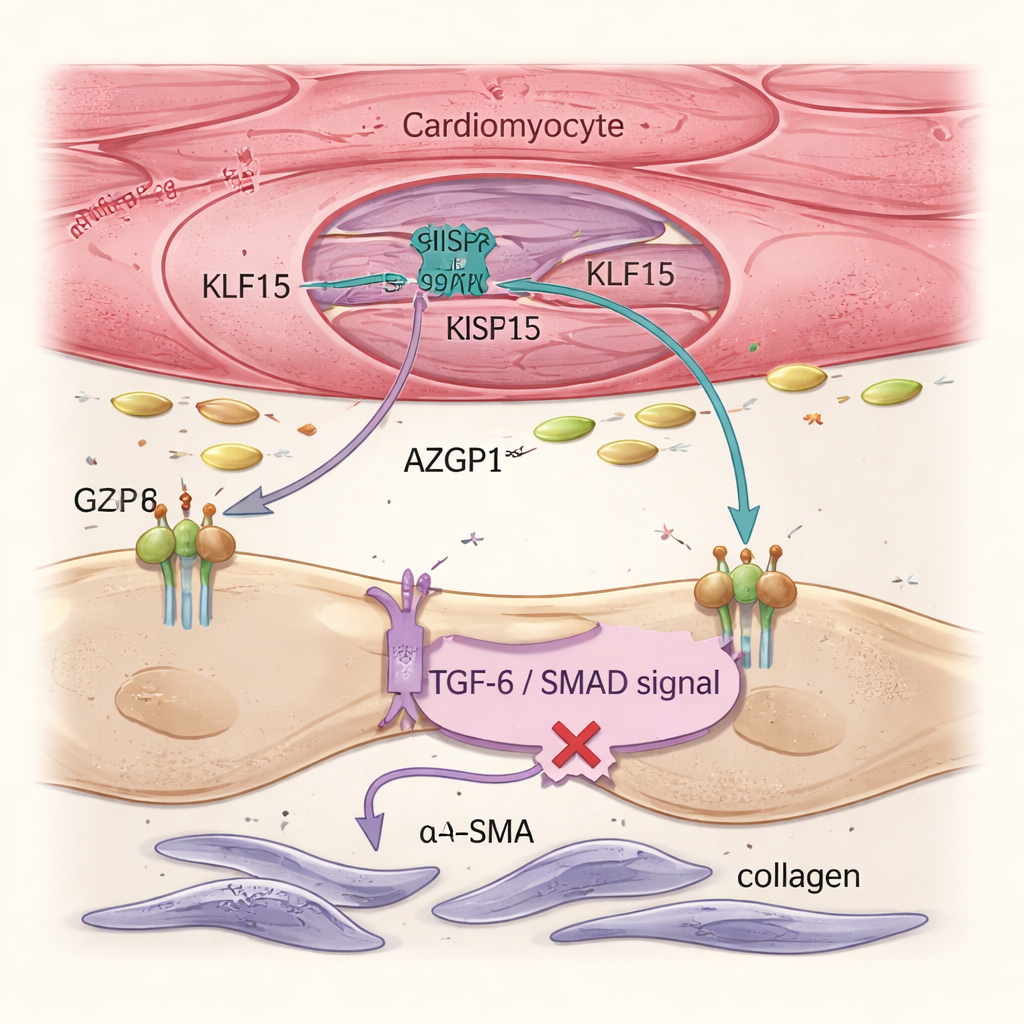
Hartfalen wordt niet alleen aangedreven door zieke spiercellen, maar ook door fibroblasten, ondersteunende cellen die collageen produceren en stijf littekenweefsel vormen. Single-cell-analyses en weefselbeeldvorming toonden aan dat het herstellen van Klf15 in cardiomyocyten de activatie van fibroblasten en de algehele fibrose verminderde, hoewel de gentherapie fibroblasten nooit rechtstreeks targette. Het team tracerde dit effect naar een uitgescheiden eiwit genaamd AZGP1. Wanneer Klf15 in cardiomyocyten werd opgevoerd, nam de productie en afgifte van AZGP1 toe. In zowel muizenharten als in menselijk stamcel-afgeleide hartweefsels dempte hogere AZGP1 de TGF-β / SMAD-route in fibroblasten — een belangrijke aanjager van littekenvorming — en verlaagde niveaus van markers zoals α-SMA en POSTN. Belangrijk is dat overexpressie van AZGP1 alleen in cardiomyocyten de spiercellen niet herprogrammeerde, wat laat zien dat KLF15 primair cardiomyocyten direct beschermt en AZGP1 gebruikt als boodschapper om fibroblasten te remmen.
Menselijke weefsels bevestigen de beschermende schakeling
Om te testen of deze mechanismen ook in menselijke cellen gelden, gebruikten de onderzoekers geïnduceerde pluripotente stamcel-afgeleide cardiomyocyten gekweekt in driedimensionale geengineerde hartweefsels. Wanneer deze weefsels mechanische belasting ondergingen die hoge bloeddruk nabootst, verloren ze KLF15, schakelden stress- en foetale genen in, verstevigde het weefsel en verzwakten hun contracties — hiermee werden ziektekenmerken gerecapituleerd. Door CRISPRa-aangedreven herstel van KLF15 werd deze achteruitgang voorkomen, bleef de krachtsopwekking behouden en verschoof de genexpressie terug richting volwassen metabolisme en structuur. Gedetailleerde experimenten toonden aan dat TGF-β1, een bekende pro-fibrotische signaalstof, KLF15 in menselijke cardiomyocyten vermindert via het SMAD2/3-pad, wat helpt verklaren hoe chronische stress leidt tot maladaptieve remodelering. Ten slotte ontwierp het team een compacte, "mini" CRISPRa-oplossing gebaseerd op een kleinere Cas9-variant die in één AAV9-vector past en wordt aangedreven door een cardiomyocyt-specifieke promotor. In precisiegesneden plakjes van falend menselijk hartweefsel verhoogde deze vector succesvol KLF15-niveaus en verbeterde de contractiele prestaties gedurende dagen in cultuur.
Een blauwdruk voor zachtere gentherapie
Voor niet-specialisten is de kernboodschap dat dit werk laat zien hoe het voorzichtig opvoeren van één beschermende regelaar binnen hartspiercellen zowel hun identiteit kan stabiliseren als signalen kan afgeven die littekenvorming beperken. Door een CRISPR-gebaseerde activator te gebruiken die het DNA niet knipt, stemt de aanpak de eigen genen van het hart bij fijn af in plaats van een kunstmatig gen in te voegen. De studie definieert een TGF-β → KLF15 → AZGP1-route die mechanische stress koppelt aan schadelijke remodelering en demonstreert, in muizen, menselijke celmodellen en plakjes menselijk hartweefsel, dat het herstellen van KLF15 deze kettingreactie kan onderbreken. Hoewel nog in een preklinische fase, biedt het compacte, cardiomyocytgerichte CRISPRa-systeem dat hier wordt gepresenteerd een potentieel stappenplan voor de behandeling van veelvoorkomende, niet-genetische vormen van hartfalen door genactiviteit te herprogrammeren in plaats van het genoom te herschrijven.
Bronvermelding: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Trefwoorden: hartfalen, KLF15, CRISPR-activatie, cardiale fibrose, AZGP1