Clear Sky Science · ja
異種触媒反応性のためのエンドツーエンド・フレームワーク
なぜ触媒設計の高速化が重要か
現代社会は燃料、プラスチック、肥料、そして日常的な多くの製品を作るために触媒に依存しています。しかし、より優れた触媒を見つけることはしばしば干し草の山から針を探すような作業です。というのも、各材料は同時に数千もの微視的反応を促進し得るからです。本稿はCAREという新たな計算フレームワークを紹介します。CAREはルールベースと機械学習を組み合わせて、これら入り組んだ反応網を従来より迅速かつより完全にマッピングし、シミュレートします。これにより、クリーンなエネルギー技術や効率的な化学プロセスの指針を示すと同時に計算コストを大幅に削減することが期待されます。
混み合った反応経路の解きほぐし
固体触媒の表面では、入ってきた分子が単一の整然とした経路をたどるわけではありません。むしろ短寿命の中間体や競合する経路の迷路を通ります。従来の計算手法は人間の直感に頼って取り得る反応ステップの限られた集合を選び、量子計算でそれらのエネルギーを評価します。これは小さなネットワークでは有効ですが、系が複雑になると破綻しやすく、長期的な活性、失活、選択性を支配する稀な経路を見落としがちです。CAREは単純な構築ルールから非常に大きな反応ネットワークを自動的に生成することでこの課題に対処します。炭素・水素・酸素間のすべての妥当な結合切断・形成事象を含め、化学者が通常は除外するような経路であっても確実に取り込むよう設計されています。
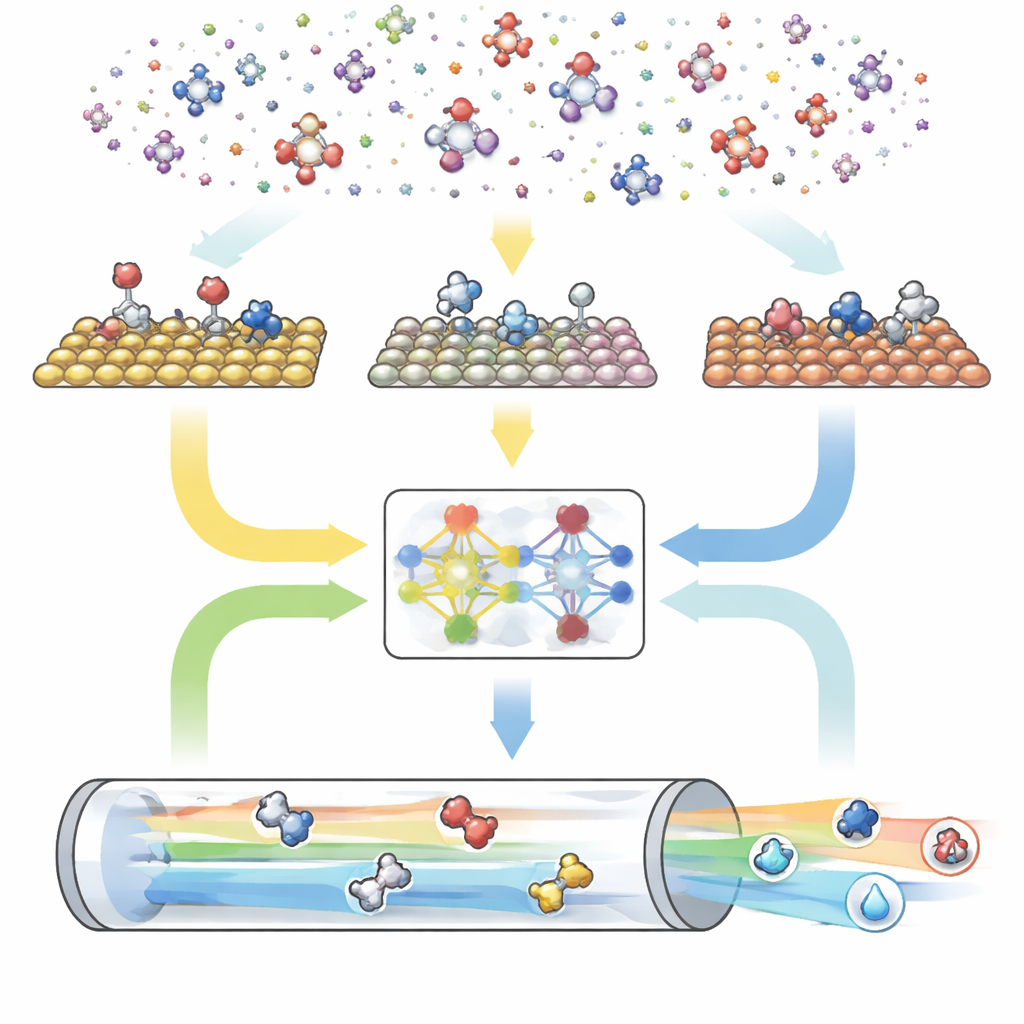
反応のための三部構成のデジタルエンジン
CAREはエンドツーエンドのパイプラインとして構築され、三つの主要モジュールで構成されます。第一に、ルールベースのジェネレーターが「化学空間」を定義します。これは炭素と酸素の最大数を設定し、単純なテンプレートを適用して一致するすべての分子と表面結合形を生成します。第二に、エネルギー評価モジュールが最新の機械学習モデル、特にGAME-Net-UQと呼ばれるグラフニューラルネットワークを呼び出して、多くの金属表面上での中間体と遷移状態のエネルギーを推定します。このモデルは各構造を原子と結合のネットワークとして扱い、エネルギーと不確かさの両方を返し、精度は数十分の一電子ボルトの範囲でありながら軽量で高速です。第三に、マイクロキネティックソルバーがこれらのエネルギーを用いて温度、圧力、電圧、pHなどの実条件下で全反応がどのように進行するかを計算し、全体の反応速度、表面被覆率、生成物の選択性を予測します。
実世界での検証:燃料分子と気候化学
CAREが単なる理論的演習にとどまらないことを示すために、著者らは難易度が増す三つの産業的に関連する問題に適用しています。水素貯蔵に重要な反応であるメタノール分解では、控えめなネットワークを生成し、多くの金属触媒と結晶面にわたって評価します。CAREは活性のよく知られた「ボルケーノ」傾向を再現し、実験と一致してルテニウムが優れた性能を示す触媒の一つであることを正しく特定しますが、フル量子計算に必要な計算時間のごく一部で済みます。次に、銅表面上での二酸化炭素の電気化学的変換に移り、1-プロパノールやプロピレンなどの三炭素生成物がどのように生じるかに注目します。プロトンや電子、溶液条件を扱う特別なステップを含めることで、CAREはpHや印加電圧が経路をどのようにシフトさせるかを捉え、1-プロパノールがプロピレンより優先されることを正しく予測し、詳細な先行研究と整合します。
合成燃料のための巨大な反応網の探索
最も注目すべき実証はフィッシャー–トロプシュ過程から得られます。これは一酸化炭素と水素の混合気を長鎖炭化水素に変換して燃料や化学品を作るプロセスです。ここで著者らはほぼ4万種の表面種と約37万の素反応を含むネットワークを構築しており、従来の量子ベースの研究が完全に探索できる規模をはるかに超えています。CAREを用いれば、コバルト、鉄、ニッケル、ルテニウムの表面上で全中間体と主要反応障壁を標準的なハードウェアで数時間で評価でき、直接的な量子計算と比べて約百万倍の高速化を達成します。これらのネットワーク上でのマイクロキネティックシミュレーションは既知の傾向を再現します:コバルトと鉄はより長鎖の炭化水素を優先的に生成し、鉄は副反応を通じてより多くの二酸化炭素を作り、ニッケルはより強い水素化に傾くといったことです。メタン収率など一部の詳細はまだ完璧ではありませんが、このフレームワークは鎖成長を支配する結合形成ステップがどれかを明らかにし、モデルの改良が必要な箇所を浮き彫りにします。
今後の触媒研究にとっての意味
専門外の読者にとっての要点は、CAREが従来は手の届かなかった触媒表面上の巨大な反応空間を実用的に探索する手段を提供することです。ネットワーク生成を自動化し、量子エネルギーの代替として高速な機械学習“サロゲート”モデルを差し込み、得られた動力学を効率よく解くことで、候補触媒のランキング、有望な操作条件の特定、そして予期せぬ経路の発見を、より少ない人為的バイアスと計算費用で行えます。著者らは混雑した表面や溶媒効果、さらに大規模なネットワークの取り扱いといった残された課題を指摘していますが、この研究はコンピュータが二酸化炭素還元からプラスチックのリサイクル、バイオマスのアップグレードに至るまで複雑な反応を迅速にスクリーニングし、発見を試行錯誤に任せるのではなく最も有望な実験へと導く未来を示しています。
引用: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
キーワード: 異種触媒, 反応ネットワーク, 機械学習, マイクロキネティックモデリング, フィッシャー–トロプシュ合成