Clear Sky Science · ja
室温およびそれ以上での前例のない頑健性を持つ物理情報導入型原子エネルギーモデル
日常の化学にとってこれはなぜ重要か
コンピューターシミュレーションは現代の化学や材料科学の主力ツールです。高価で時間のかかる実験の代わりに、分子がひねられ、振動し、衝突する様子をシリコ上で観察できます。しかし、これらのシミュレーションが機械学習に依存すると、特に高温で突然「破綻」し、物理的にあり得ない分子形状を生成してしまうことがあります。本研究は、非常に長時間、最大1000ケルビンの温度で動作しても崩壊しない、新しいタイプの物理情報導入型機械学習モデルを紹介します。
賢い近道から壊れやすいシミュレーションへ
従来の量子化学は分子エネルギーを高精度で計算しますが非常に遅いです。より単純なフォースフィールドは高速ですが近似的です。機械学習ポテンシャルは両者の利点を組み合わせることを目指し、分子幾何からエネルギーと力への近道を学習してそれを分子動力学に利用します。一見すると、多くのモデルは標準的なテストセットで平均誤差が小さく優れているように見えます。しかし実際にはこれらの数字は誤解を招くことがあります。シミュレーション中に分子が新たな形状を探索すると、特に高温では多くのモデルが訓練範囲外へ押し出されます。現実的な形状へ穏やかに戻すのではなく、結合を伸ばしたり潰したりするような力を予測して系全体を非物理的にし、シミュレーションがクラッシュすることがあるのです。
量子に基づく構成要素に基づくモデル構築
著者らは、モデルが学習する対象と事前の物理情報の与え方を変えることでこの脆弱性に対処します。彼らはFFLUXと呼ばれる枠組みを用い、これはInteracting Quantum Atoms(IQA)アプローチに基づいています。IQAでは分子を「位相的原子」に分割し、各原子のエネルギーを量子力学から直接決定します。これらの原子エネルギーは物理的に意味があり、分子全体のエネルギーに足し合わされます。任意のサイトエネルギーを学習する代わりに、新しいガウス過程モデルはこうした量子に根ざした原子エネルギーを学習し、各予測に深い物理的根拠を与えます。ペプチドでキャップされたグリシンとセリン、マロンダルデヒド、アスピリンの四つの柔軟な有機分子が、多くの内部運動を持ち、既存の機械学習フォースフィールドにとって難しい試験ケースとして用いられました。
問題を予期するようにモデルを教える
重要な革新は、ガウス過程がデータを見る前にどのように設定されるか、すなわち「平均関数」にあります。これはモデルが情報の乏しい領域で何を期待するかを符号化します。これまでの多くの研究はこの平均をゼロに設定し、事実上事前期待がないふりをしてきました。著者らは代わりに、この平均を物理的に妥当な範囲でより高い原子エネルギー状態側へ意図的にシフトさせました。この設計により、モデルが外挿を余儀なくされる場面—例えば結合が一時的に過度に伸びたとき—には極端な歪みを罰する方向の予測を自然に好むようになります。広範なテストで、事前分布だけが異なるモデル群は非常に異なる振る舞いを示しました。従来型や低エネルギーの平均を持つモデルは、多くの場合ピコ秒未満で分子が爆発または崩壊してしまいました。対照的に、最良の高エネルギー平均(MF5と名付けられた)は、300〜1000ケルビンの温度範囲で四つの分子すべてに対して、ナノ秒単位の試験ウィンドウ全体で安定したシミュレーションをもたらしました。
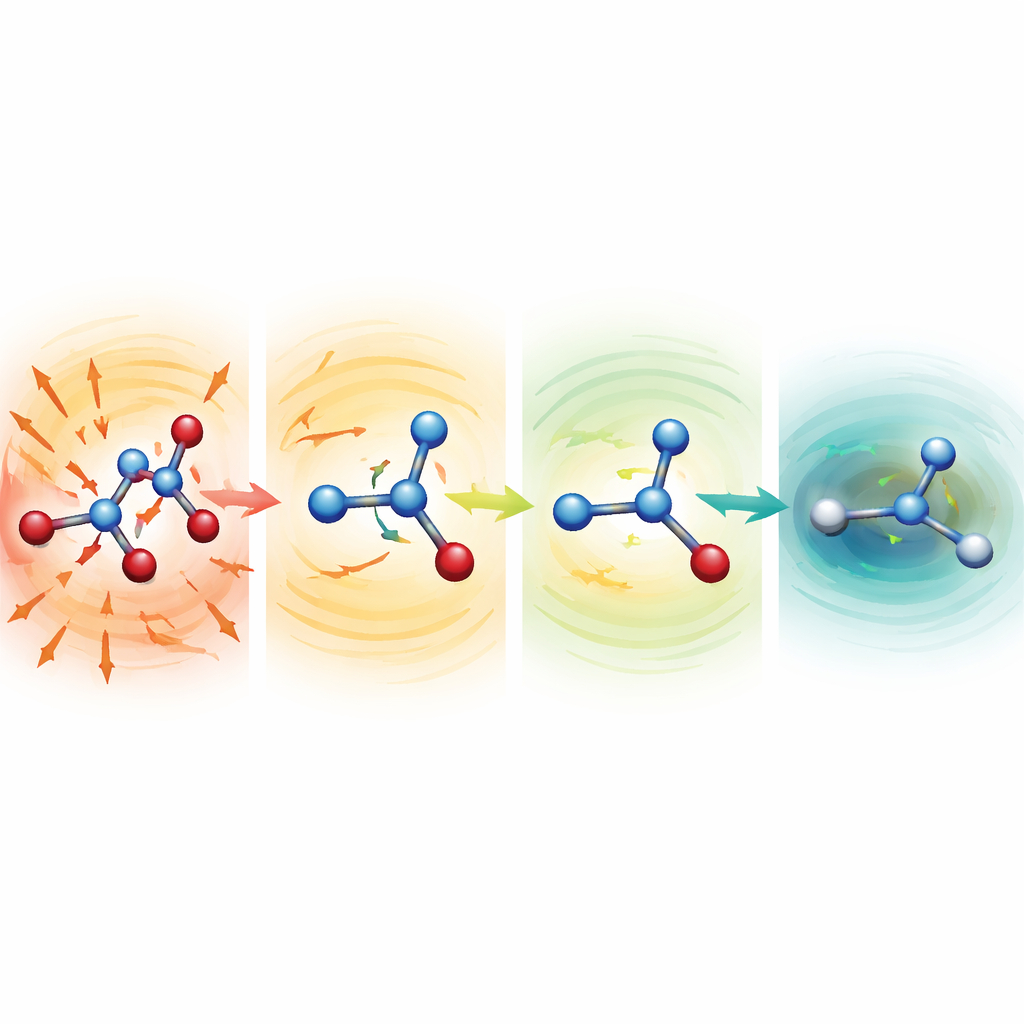
歪んだ分子が自己修復する様子を観察する
頑健なモデルがうまく機能する理由を調べるため、研究者たちは意図的にひどく伸長あるいは圧縮された結合を持つ毀損構造からシミュレーションを開始しました。セリン、アスピリン、マロンダルデヒドでは、これらの出発点は通常の構造より数百から千キロカロリー以上も高いエネルギーに相当し、通常なら破滅的です。弱い平均関数を持つ場合、分子はすぐに引き裂かれてしまいました。しかしMF5の設定では、予測された力は即座に伸びた結合を縮め、潰れた結合を伸ばす方向を指しました。数十~数百ステップのうちに分子は現実的な形状へと緩和し、その後も安定に進化し続けました。さらに、力を一度も教え込んでいないにもかかわらず、同じモデルがアラニンジペプチドの幾何最適化を導き、既知の低エネルギー配座と相対エネルギーを数十分の一キロカロリー程度の精度で再現しました。しかも完全な量子計算と比べて約200倍低いコストで実行できました。
汎用的なハードウェアでの長時間・高温シミュレーション
頑健性とは一度の困難な開始を生き延びることだけではなく、数百万から数十億ステップにわたって持ちこたえることです。著者らは最良モデルをさらに追試し、500ケルビンでそれぞれ10ナノ秒の独立したシミュレーションを四つの試験分子について50本実行しました。これらのランでは一度もクラッシュせず、合計で半マイクロ秒のシミュレーション時間を達成しました。これは最先端の機械学習フォースフィールドとしては異例です。さらに注目すべきは、シミュレーションが標準的なCPUで効率的に実行され、ステップごとの速度が高性能GPUを必要とするいくつかの著名なニューラルネットワークポテンシャルに匹敵するかそれを上回った点です。シミュレーション中、分子は豊富な形状や準安定状態を探索しており、頑健性が運動を人工的に固定したり剛直な構造を強制した結果ではないことが示されました。
将来の分子モデリングにとっての意義
非専門家に向けた要点は、低誤差を示す機械学習モデルのすべてが、強く押し込まれたときに信頼できるわけではないということです。量子由来の原子エネルギーにモデルを根ざし、モデルの内蔵期待値を高エネルギー側へ慎重に調整することで、著者らは「復元力」—分子における安全帯のようなもの—を自然に生み出す一群のポテンシャルを作り出しました。それにより、高温や歪んだ初期状態でもシミュレーションを物理的に保つことができます。このアプローチは複雑な分子のより信頼できる長時間シミュレーションを約束し、今後は凝縮相や分散など微妙な相互作用を扱う同様の物理情報導入型モデルへの拡張が期待されます。しかも計算実用性を維持したまま実現できます。
引用: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
キーワード: 機械学習フォースフィールド, 分子動力学の頑健性, ガウス過程ポテンシャル, 物理情報を取り入れたモデリング, 量子的原子エネルギー