Clear Sky Science · ja
解釈可能な機械学習がコンフォーマル予測と反事実説明により炭酸脱水酵素阻害を合理化する
より賢いがん薬が重要な理由
抗がん薬はしばしば粗雑な道具のように振る舞います:腫瘍細胞を攻撃する一方で、健康な組織にも作用し重篤な副作用を引き起こすことがあります。これを改善する有望な手段の一つは、低酸素環境で腫瘍の生存を助ける炭酸脱水酵素の特定のアイソフォームを阻害することです。しかしこの酵素のいくつかの形態はほとんど同一に見えるため、腫瘍の「悪い」アイソフォームだけを標的にし、全身に存在する「良い」アイソフォームを損なわない薬を設計するのは困難です。本研究は、解釈可能な機械学習がこの課題をどう扱い、より選択性が高く安全な候補薬を設計するのに役立つかを示しています。
誤った標的を叩く問題
ヒトの炭酸脱水酵素(hCA)には多くのアイソフォームが存在します。そのうちIXとXIIは低酸素の腫瘍でがん細胞の生存に関係しており、これらを阻害すれば病勢を抑え治療効果を高める可能性があります。しかしアイソフォームIIは健康な組織で広く発現しており、その活性部位はIXやXIIと非常によく似ています。これら三つすべてに結合する薬は、代謝性アシドーシスや視覚障害などの望ましくない問題を引き起こす可能性があります。酵素は大きく複雑な分子であり、薬様化合物の組合せは天文学的に多いため、従来の実験や計算手法では全てを網羅的に試すことは事実上不可能です。
クリーンで信頼できるデータ基盤の構築
著者らはまず、ChEMBLリポジトリからhCA II、IX、XIIに対して試験された数千の分子を慎重にクリーンアップしたデータベースを組み立てることから取り組みました。化学構造を標準化し、疑わしい測定値を除外し、この阻害剤クラスに典型的な亜鉛結合基を共有する化合物に注目しました。厳格なカットオフを用いて分子を明確に活性または非活性とラベル付けし、モデルを混乱させる境界ケースは除外しました。非活性が活性より遥かに多かったため、学習アルゴリズムが多数派クラスを単に好むことがないようデータのバランスを取っています。さらに、訓練セットとテストセットが異なるコア骨格を含むようスキャフォールド(骨格)ベースの分割を用い、モデルが真に新しい化合物に対してどの程度うまく扱えるかを現実的に評価しました。
データが限られるときは単純なモデルが深層学習を上回る
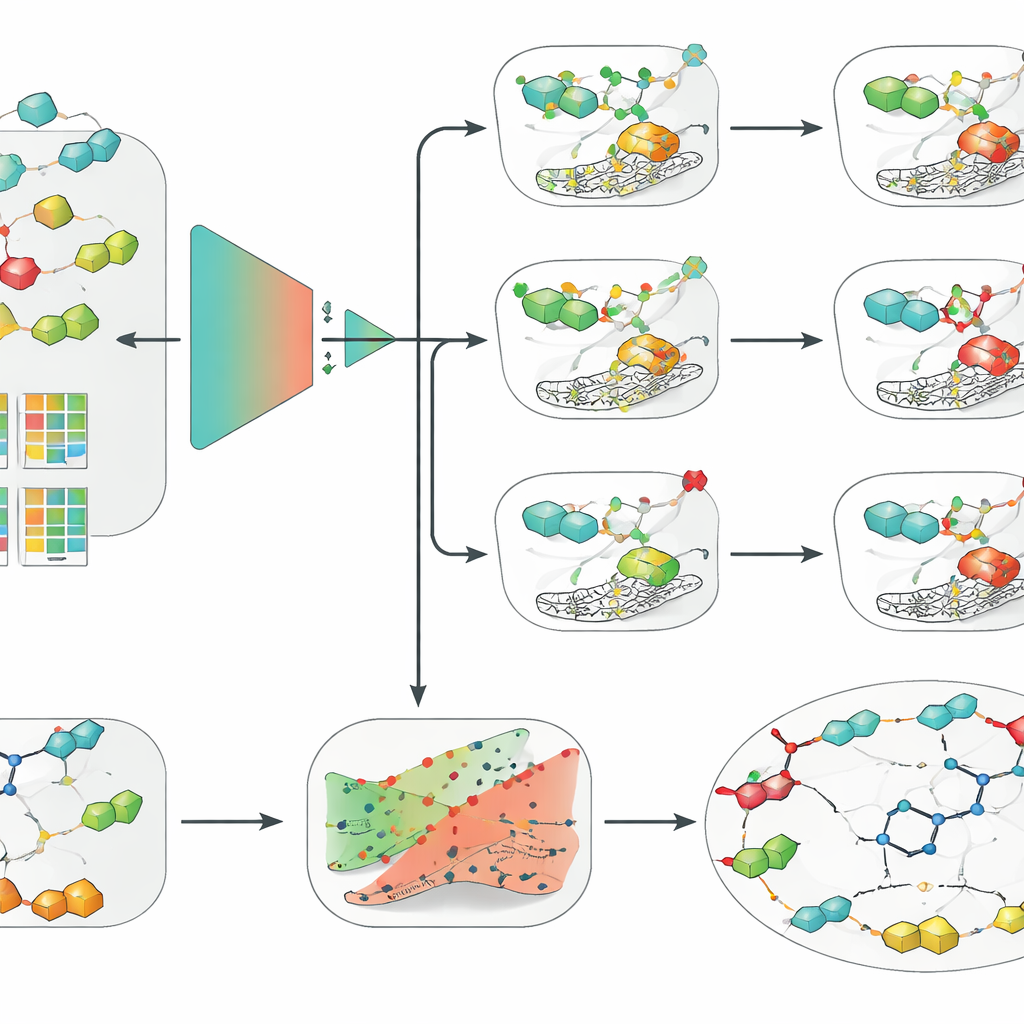
この精選されたデータセットに対して、研究チームはロジスティック回帰、ランダムフォレスト、サポートベクターマシン(SVM)といった古典的機械学習手法から、分子構造に直接作用するグラフベースのモデルを含む現代的な深層ニューラルネットワークまで幅広い手法を比較しました。分子の符号化方法としては、従来の手作り記述子、キー型フィンガープリント、化学言語モデルから学習された埋め込みなどを組み合わせました。三つのアイソフォーム全てにおいて、厳格なスキャフォールドベース評価下で一貫して優れていたのは、拡張結合性フィンガープリント(ECFP)を入力としたSVMの組合せでした。意外にも、この比較的単純な構成がグラフや深層学習の流行モデルを上回り、データ品質、厳密な検証、良い分子記述子が、データセットが中程度のサイズである場合にはアルゴリズムの複雑さより重要になりうることを強調しています。
信頼できる確信度と人に優しい説明の付与
研究者らは最良のSVMモデルに、実際の創薬で使いやすくするための二つの層を追加しました。まず、コンフォーマル予測というフレームワークを適用しました。これは単なる二値答えを出すのではなく、許容された誤り率を保証しつつ、ありうる結果の領域を示します。これにより科学者はモデルの慎重さを調整でき、モデルが本当に不確かであるケースを認識できます。次に、反事実説明を用いてモデルの推論を直感的にしました。ある分子に対して、予測を活性から非活性(またはその逆)に反転させるような非常に近縁の類縁体を生成します。臨床候補物質SLC-0111(IXとXIIを選択的に阻害しIIは阻害しない)についてこれらの対を調べると、本手法は重要なメディシナルケミストリーの知見を独立に再発見しました:分子の「テール」部分の小さな変化が、どのアイソフォームに好んで結合するかを強く変えるという点です。
アルゴリズムから実践的な創薬ツールへ
アプローチを利用しやすくするために、著者らは三つのSVMモデル、不確実性層、反事実エンジンを統合したグラフィカルツール「CAInsight」をパッケージ化しました。ユーザーは分子のテキスト表現を入力すると、ワンクリックでhCA II、IX、XIIに対する予測活性、それぞれの予測の信頼度推定、および活性を高めたり下げたりする可能性のある構造的修正案を得られます。これらのモデルは一歩で正確な効力や選択性を予測するというよりは、活性/非活性を分類することに焦点を当てていますが、実際の候補薬について既知の挙動を再現し、微妙な構造変化を区別できています。著者らは、より大きく均質なデータセットや活性カットオフの選び方に関するより深い解析が性能をさらに改善し得ると指摘しています。
将来のがん治療薬にとっての意義
平たく言えば、本研究は慎重に構築され説明可能な機械学習モデルが、外見の似た酵素標的をよりよく区別するがん薬の設計を化学者に助け得ることを示しています。堅牢な統計、不確実性の推定、直感的な「もし〜なら」例を組み合わせることで、このフレームワークはどの分子が有望かを予測するだけでなく、その理由も示唆します。こうした透明性のある人工知能はバーチャルスクリーニングを加速し、新規化合物の生成設計を支援し、実験室での試行錯誤の負担を減らすことで、最終的にはより選択的で安全な治療法の発見に寄与する可能性があります。
引用: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
キーワード: 炭酸脱水酵素阻害剤, 解釈可能な機械学習, 薬物選択性, コンフォーマル予測, 反事実説明