Clear Sky Science · ja
高次元空間向けヒープ駆動型進化フレームワークによるがんマイクロアレイデータの特徴選択最適化
正しい遺伝子を選ぶことが重要な理由
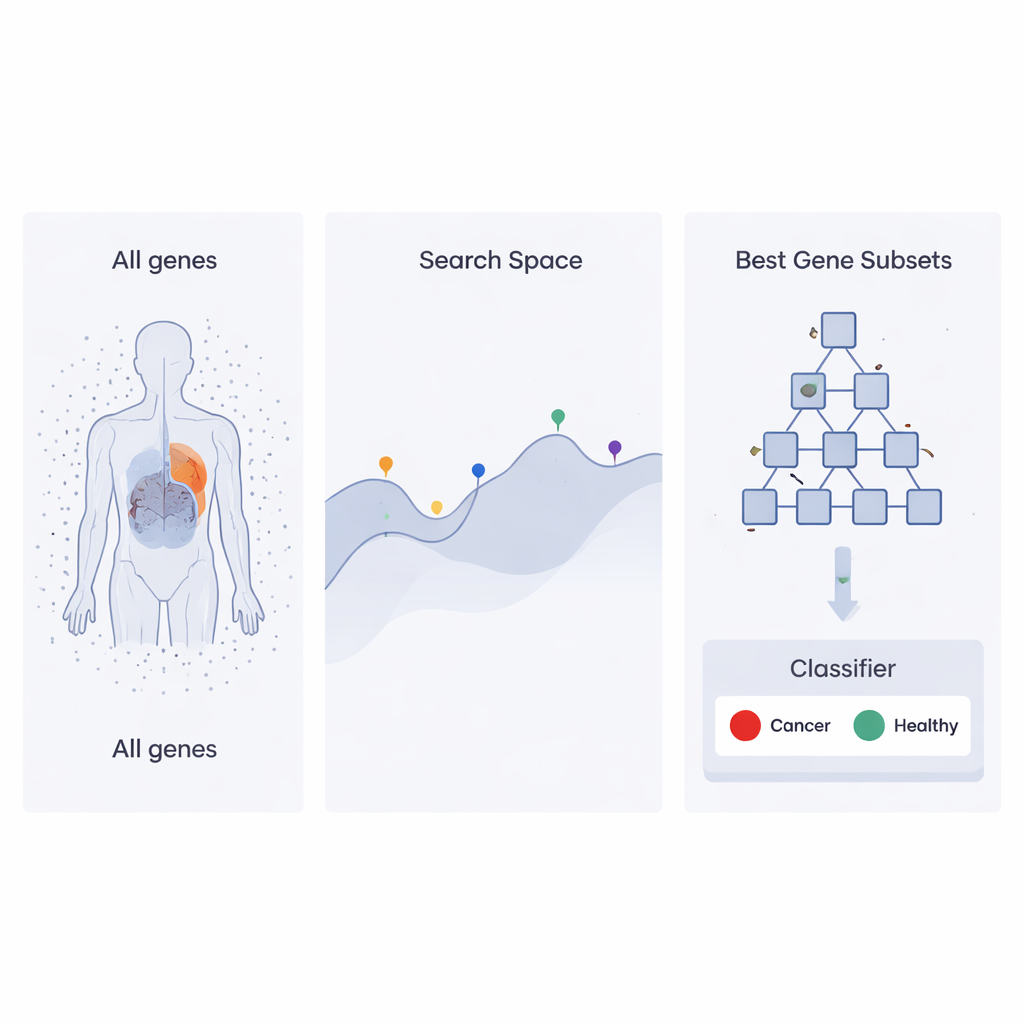
現代の遺伝学的技術を用いたがん検査は一度に数万の遺伝子を測定できますが、臨床では数十例分のサンプルしか得られないことが多いです。この膨大な「遺伝子のジャングル」の中には、ごく少数のシグナルだけががんのタイプ間や腫瘍と正常組織を確実に区別します。本論文は、その重要な遺伝子を自動的に選び出す新しい賢い探索手法を提案し、コンピュータ支援によるがん診断をより正確で、より高速かつ解釈しやすくすることを目指しています。
シグナルは多すぎ、データは少なすぎる
マイクロアレイ実験や類似の技術は、各患者サンプルについて数千の遺伝子の発現レベルを測定できます。一方でサンプル数は通常非常に限られ、場合によっては百件未満です。これらの遺伝子測定値の多くはノイズが多く、冗長であったり対象の疾患に無関係だったりします。全てを保持すると学習アルゴリズムが圧倒され、計算が遅くなり、真の生物学的特徴ではなく偶発的な特異点に引きずられる誤ったモデルが生まれることがあります。このようなデータを有用な部分集合に削減する過程を「特徴選択」と呼び、高次元の医療データから信頼できる予測を得るために極めて重要です。
企業の組織図に着想を得た探索戦略
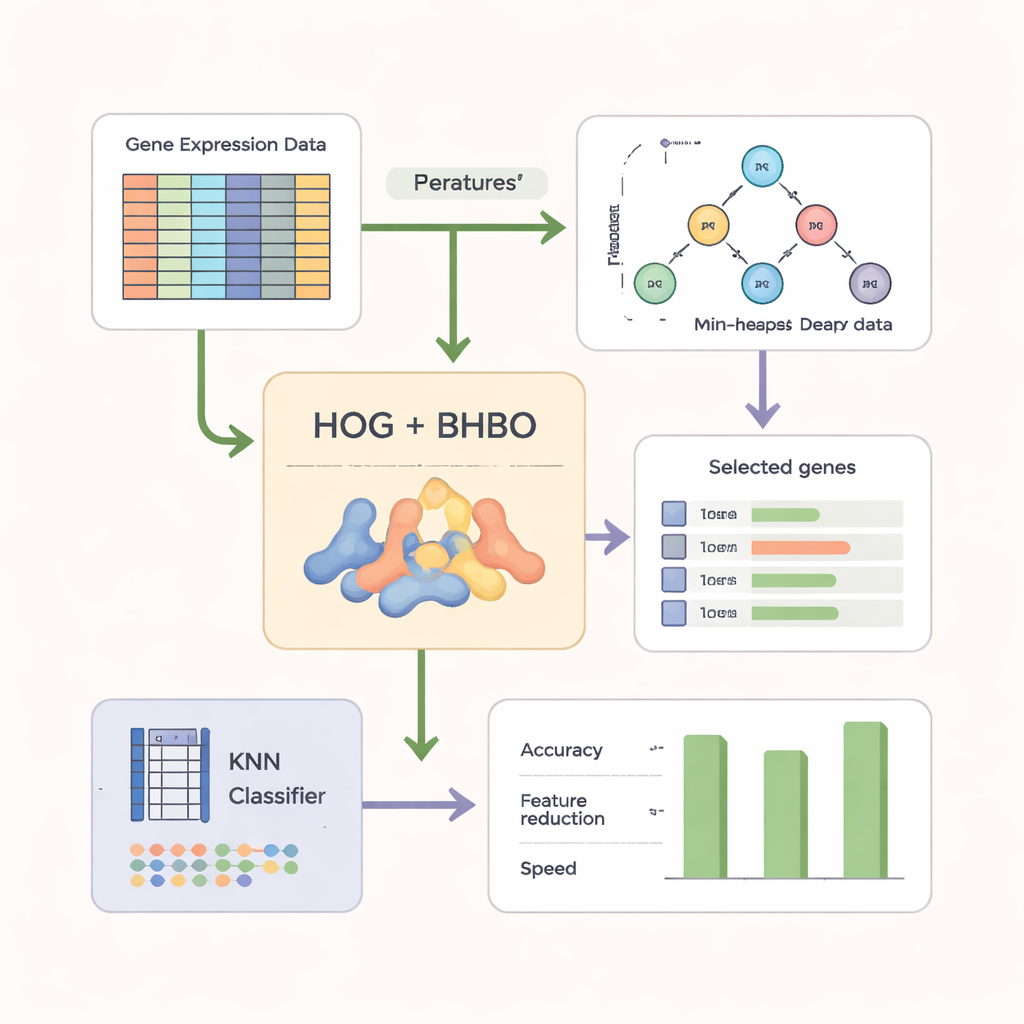
著者らはHeap‑Based Optimizer(HBO)と呼ばれる最近の最適化手法を発展させています。これは社員の組織化の仕方に着想を得たものです。各遺伝子集合を、それが分類器によりがんサンプルと正常を区別するのにどれだけ寄与するかで評価される「社員」とみなします。これらの社員はヒープと呼ばれるコンピュータ構造を使って階層的に並べられます。高性能な遺伝子集合は上位に、性能の低いものは下位に配置されます。何度も繰り返すうちに、下位の個体は上位や同僚の振る舞いを模倣し少しずつ改変することで、自らの選択を調整し、組織全体をより良い解へと徐々に導きます。
生の遺伝子データをより鋭いパターンへ変換
探索をより効果的にするために、著者らは生の遺伝子測定値だけに依存しません。まずマイクロアレイデータを画像のような形式に再構成し、コンピュータビジョンで広く使われるHistogram of Oriented Gradients(HOG)という手法を適用します。HOGは遺伝子間での発現レベルの変化を捉え、単独の測定値よりも局所的なパターンを強調します。これらのパターンベースの特徴は元の遺伝子情報と組み合わせられます。単純な分類器であるk‑近傍法(KNN)が「審判」として機能し、新しいサンプルに対するラベリング精度で各候補遺伝子集合を評価すると同時に、より小さく凝縮された集合を評価するように報酬を与えます。
複数のがんデータセットでの検証
研究者たちは、Heap‑Based Optimizerの二項版(BHBO)を脳腫瘍、白血病、前立腺がん、亜型の多い混合腫瘍コレクションなど、9つの公開がんマイクロアレイデータセットで評価しました。各データセットは数千〜1万5千を超える遺伝子を含む一方で患者サンプル数は比較的少数でした。各データセットについてBHBOを何度も実行し、遺伝的アルゴリズムや粒子群最適化などのよく知られた7つの探索手法と比較しました。評価は単に精度だけでなく、保持された遺伝子数、探索の収束速度、ノイズ、バッチ効果、ラベル誤りなどを模擬したデータ攪乱に対する結果の安定性も測っています。
新手法が達成したこと
9つのデータセット全体で、ヒープ駆動アプローチは平均約95%の分類精度を達成しつつ、遺伝子数を85%以上削減しました。いくつかのデータセットでは競合手法を明確に上回り、探索の収束も速く—つまりより少ない探索ステップで良好な遺伝子集合に到達しました。著者らが意図的にデータを汚損(ノイズの追加やサンプルラベルの反転)しても、手法の性能低下はわずかで、代替手法より高い性能を維持しました。統計検定により、これらの改善が偶然による可能性は低いことも示されています。
将来のがん診断にとっての意義
実務的には、本研究は巧妙に設計された探索戦略が膨大な遺伝子データから情報量の高い小さな遺伝子パネルを抽出し、それでも高い分類性能を保てることを示しています。臨床医や研究者にとって、そのような凝縮された遺伝子集合は生物学的な検証が容易で、追跡検査のコストが低く、意思決定支援ツールへの組み込みにも適しています。本手法は直接新薬や経路を発見するわけではありませんが、有望な遺伝子マーカーに注目を集め、高次元のがんデータに潜む最も情報量の高いシグナルに他の研究が集中できるよう助けます。
引用: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
キーワード: がんマイクロアレイ, 特徴選択, メタヒューリスティック最適化, 遺伝子バイオマーカー, 医療データマイニング