Clear Sky Science · ja
極めて高次元のゲノムデータのための確率的LASSO
ゲノムの干し草の山から針を見つける
現代の生物学では数万もの遺伝子を同時に測定できますが、患者研究ではしばしば数百人程度しか含まれません。この不均衡の中に、疾患リスクや生存予後の予測に本当に重要なごく少数の遺伝子群が隠れています。本論文は「確率的LASSO」と呼ばれる統計手法を紹介します。これは、患者数より遺伝子数がはるかに多い場合でも、膨大でノイズの多いゲノムデータから重要な遺伝子を確実に見つけ出すよう設計されています。
適切な遺伝子選択が難しい理由
研究者はしばしば、重要でない遺伝子の影響をゼロへ縮小しながら情報量の多い遺伝子を残すLASSOのようなツールに頼ります。しかし、クラシックなLASSOは、がんゲノミクスで一般的なように遺伝子数がサンプル数を圧倒する場合に苦戦します。標準的なLASSOは最大でも患者数と同じ数の遺伝子しか選べず、類似した振る舞いを示す遺伝子を見落としがちです。相関を扱うために追加のペナルティを加える改良手法もありますが、それらは関連する遺伝子をすべて同じ方向に結果を押しやるかのように扱ってしまい、生物学的な意味合いをぼやけさせることがあります。
より「きれいな」ランダムサンプルの作成
有望な回避策の一つは、より小さな遺伝子のランダムサブセットに対してLASSOを繰り返し適用し、その結果を組み合わせる方法です。しかし、こうした「ブートストラップ」アプローチには三つの問題があります:相関する遺伝子が互いに相殺してしまうこと、多くの遺伝子がめったにまたは全くサンプリングされないこと、そして純粋なランダム性が最終的な選択を不安定にすること。確率的LASSOは相関ベースのブートストラップと呼ぶ新しいサンプリング方式でこれらの問題に正面から取り組みます。遺伝子を単にランダムに選ぶ代わりに、既に選ばれた遺伝子とあまり相関しない遺伝子を意図的に優先して選び、より独立性の高い小さな遺伝子集合を作ります。また、全ブートストラップ実行中に各遺伝子が同じ回数だけ使われるようにして、ある遺伝子が不当に無視されないようにしています。 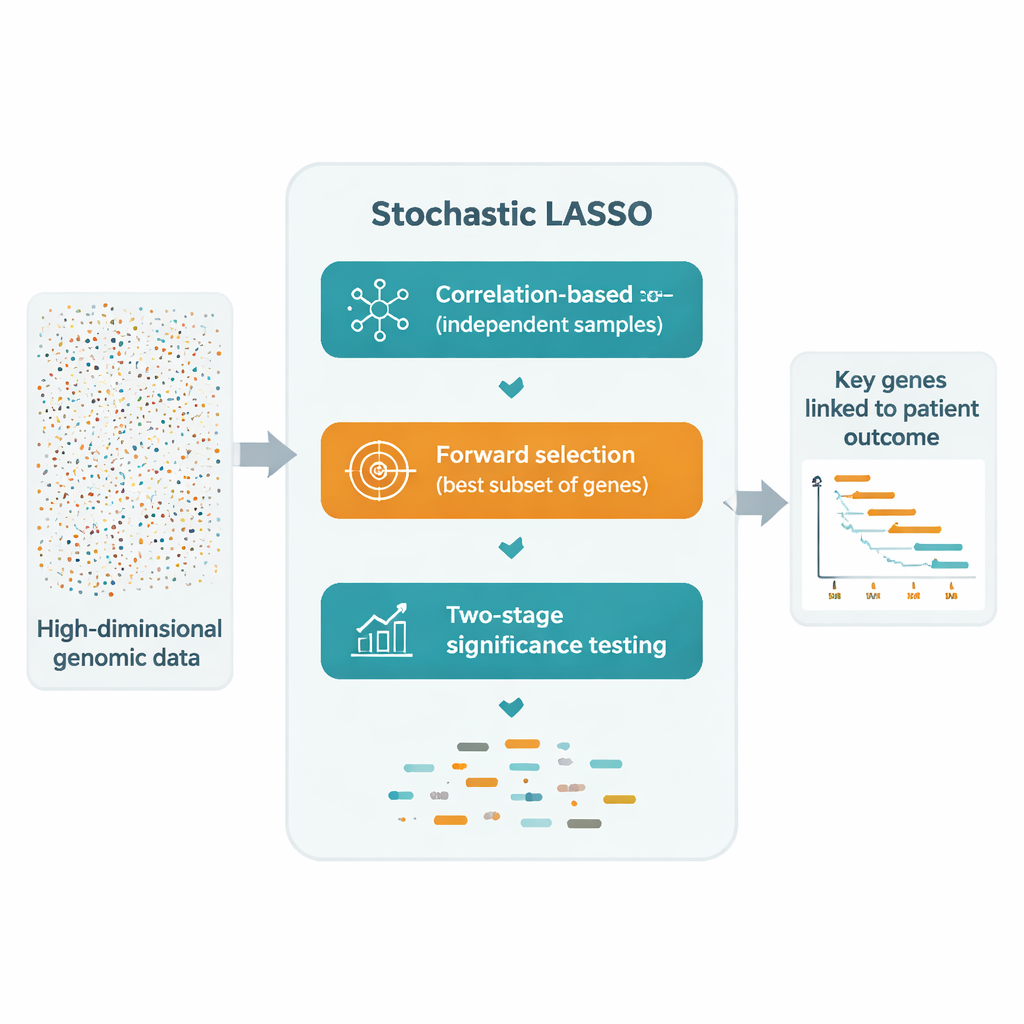
局所的な手がかりからグローバルな遺伝子集合へ
こうして作られたより「きれいな」サブセットの後、確率的LASSOはブートストラップごとの各遺伝子の係数の大きさを記録します。この平均絶対効果が、その遺伝子がどれだけ一貫して重要かを示す「局所スコア」になります。全ての可能な組み合わせを総当たりで試す代わりに、方法は局所スコアの順に遺伝子を追加して候補モデルを構築し、それぞれの候補が別の検証データでどれだけ予測できるかを評価します。こうして、従来の逐次探索法よりはるかに少ない試行で、データを最もよく説明するコンパクトな遺伝子集合を決定します。
どの遺伝子が本当に重要かの検証
「頻繁に選ばれる」から「統計的に説得力がある」へ進めるために、著者らは二段階のt検定を導入します。まず各遺伝子のブートストラップ平均係数がゼロと明確に異なるかを確認し、潜在的に意味のあるものとしてフラグを立てます。次に、これらの候補の中で各遺伝子の効果が候補全体の典型的な効果サイズより大きいかを検討します。両方の検定を通過した遺伝子のみが有意と宣言されます。これらの検定は多数のブートストラップ推定に依拠するため、確率的LASSOは従来のLASSOでは不可能な、患者数を超える数の有意遺伝子を自信を持って特定できます。 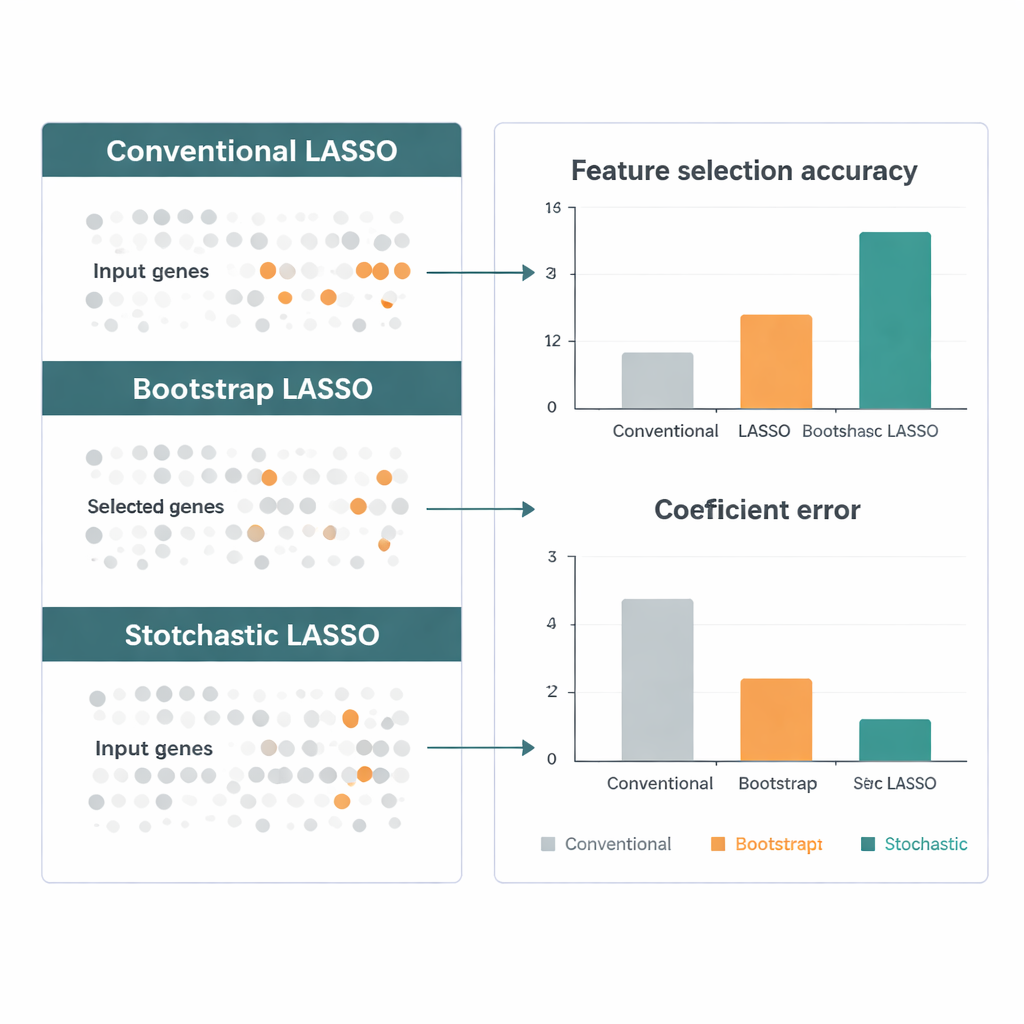
シミュレーションとがんデータでの有効性の実証
著者らは、実際のゲノム研究を模した非常に多数の遺伝子、強い相関、既知の「真の」信号を含むシミュレーションデータを用いて、確率的LASSOを複数の主要なLASSO変種と比較ベンチマークしました。複数のシナリオにわたり、新手法は正しい遺伝子をより頻繁に見つけ、効果量をより正確に推定し、実行ごとの安定性も保ちました。次に、脳腫瘍、特に悪性のグリオブラストーマを含むThe Cancer Genome Atlasの遺伝子発現データに適用しました。確率的LASSOは患者生存に関連する数百の遺伝子を浮き彫りにし、シグナル伝達や薬物代謝経路など文献上の独立した支持を持つ生物学的経路も示唆しました。これは統計的に鋭いだけでなく生物学的にも妥当であることを示唆します。
患者と研究者にとっての意義
専門外の読者にとっての要点は、確率的LASSOがゲノム大規模データのためのより賢いフィルターであるということです。限られたデータや高度に相互連結した遺伝子群の下でも、真の疾患関連遺伝子を統計的ノイズから分離するのに役立ちます。より正確で安定した遺伝子リストと効果推定を提供することで、がんや他の複雑な疾患におけるバイオマーカー、薬剤標的、予後指標の探索を鋭くします。線形回帰で実証されていますが、同じフレームワークは生存解析モデルや分類問題にも組み込むことができ、生物医学研究全般にわたる潜在的な影響を広げ得ます。
引用: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
キーワード: ゲノム特徴選択, 高次元データ, LASSO法, がん遺伝子発現, バイオマーカー探索