Clear Sky Science · ja
パンデミック規模の系統解析における進化速度の変動と再発する配列エラー
今後の流行にとってなぜ重要か
新しいウイルスが世界中に広がると、研究者はその遺伝コードを読み、系統樹を再構築することを急ぎます。系統樹は変異株の発生や広がりの速さ、対策の効果を追跡するのに役立ちます。しかしCOVID-19では、研究室がSARS‑CoV‑2のゲノムを急速に数百万件も配列決定したため、データに潜むエラーや特異性が図を歪め始めました。本論文は、そのような膨大な遺伝データを精査・解釈する新たな手法を示し、パンデミックウイルスが実際にどのように進化し集団内を移動するかをより明瞭に示すことを可能にします。
何百万のゲノムを解釈する難しさ
ゲノム疫学はウイルスゲノムを公衆衛生の実用的情報に変えます。SARS‑CoV‑2では、全世界で2,000万件以上のゲノムが共有されました。従来の進化解析ツールは種間の遺伝子比較など、より控えめな問題を想定して作られており、リアルタイムで到着する何百万ものほぼ同一なウイルス配列を扱うことは想定外です。この規模では、特に二つの問題が厄介になります。第一に、ウイルスゲノムの一部の塩基位置は他よりもはるかに頻繁に変異し、系統的に無関係なウイルスが奇妙に似て見えることがあります。第二に、シーケンシングやデータ処理における繰り返し発生する技術的エラーが実際の変異を模倣することがあります。これら両方の効果は系統樹に「偽の反響」を生み、どの枝やグループを信頼すべきかについて不確実性を生じさせます。
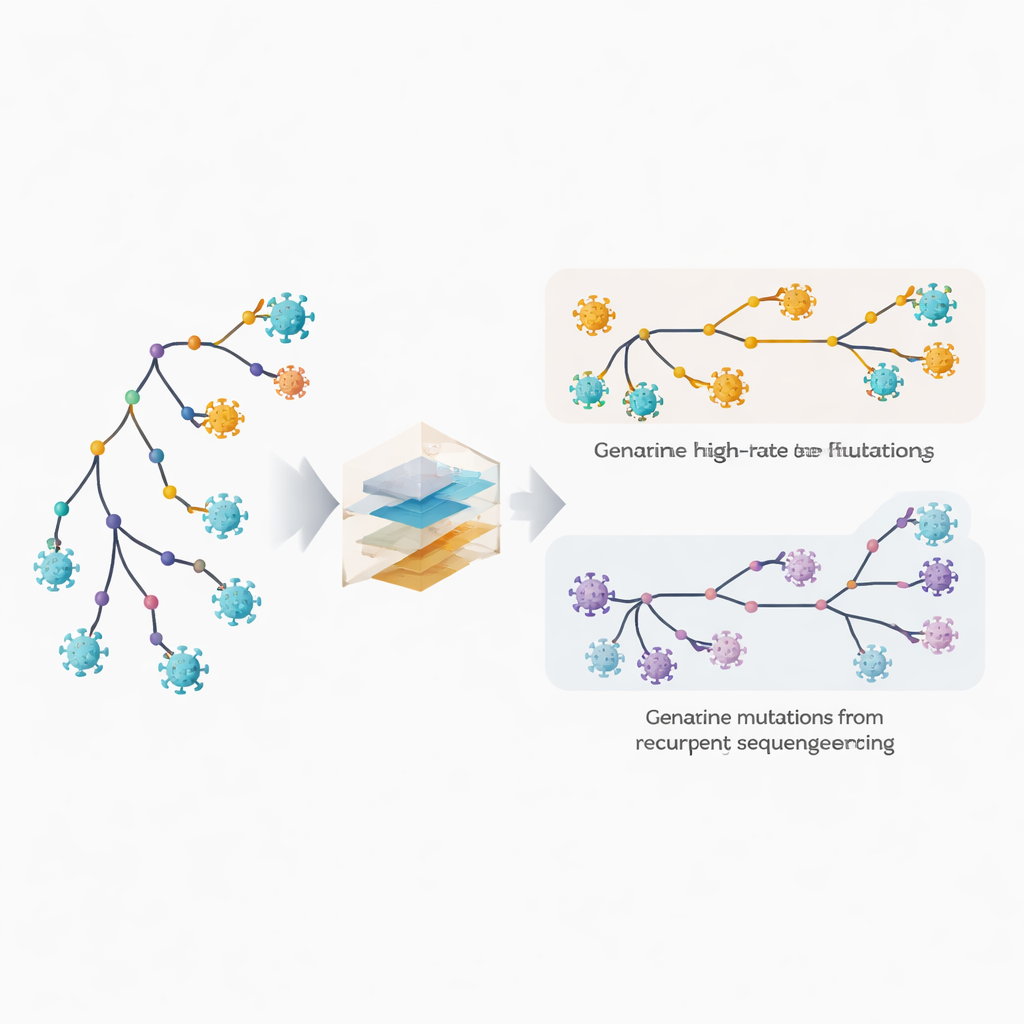
変化の速い部位と隠れた誤りを見つける
著者らは系統解析ソフトウェアMAPLEを拡張し、ウイルスゲノムの各位置がそれぞれ固有の振る舞いを持つとみなすモデルを導入しました。平均的な変異率の数値を仮定する代わりに、膨大な数のゲノムを利用して各塩基位置ごとに個別のレートを推定します。同時に、各位置が繰り返し起きるシーケンシングやコンセンサス決定のエラーを生じやすい確率も持てるようにしています。鍵となる手法は、変化が系統樹の内部の深い枝(より古く共有された事象を反映する)にどれだけ現れるかと、外側の末端(個々のゲノムに対応する)にどれだけ現れるかを比較することです。真の生物学的変異は内部枝と末端枝の間に分散しやすいのに対し、技術的エラーは主に末端に現れます。このパターンを利用することで、本当に速い進化と繰り返す誤りを判別できます。
密集した系統樹を扱うための高速アルゴリズム
何百万というゲノムを扱うには通常、膨大な計算資源が必要です。解析を現実的にするために、チームはMAPLEが系統樹上で配列情報を格納・更新する方法を再設計しました。すべてのゲノムを単一の固定参照と比較する代わりに、ソフトウェアは系統樹内部の「局所参照」点を選び、近傍のゲノムをこれらのアンカーに対する差分として記録します。このコンパクトな表現は系統樹の離れた部分同士の比較を高速化します。さらに、新しいサンプルを既存の樹に追加する方法、枝長の調整、代替的な樹形の探索の効率化などが改良され、最も負荷の高い処理は複数のプロセッサコアに並列実行するオプションもあります。
手法の検証と実データの精製
モデルが機能するか確認するために、著者らはまず既知の変異パターンと埋め込まれた配列エラーを持つ現実的なSARS‑CoV‑2のシミュレーションデータセットを作成しました。これらのテストでは、新しいアプローチはより真の系統樹を復元し、特に数万件以上のゲノムを含む場合に個々のエラーを高精度で特定しました。次に実データに移り、生のリードが利用可能な数百万件のSARS‑CoV‑2配列を解析しました。二つの異なるコンセンサス構築パイプラインを比較することで、プライマー結合の問題や参照バイアスによる呼び出しなど、人工的な影響を繰り返し受ける特定のゲノム位置を突き止めました。これらの疑わしい部位はさらなる解析からマスクされ、汚染や混合感染の兆候を示すゲノムは除外され、200万件を超える高品質配列の精選アライメントが得られました。
ウイルスの系統樹のより明瞭な世界像
精査されたデータセットを用いて、著者らは世界規模のSARS‑CoV‑2系統樹を再構築し、主要変異株同士の関係をマッピングしました。彼らの樹は、しばしば以前の公開系統樹とは微妙に異なる関係を示し、それらは多くの場合より少ない変異イベントで説明でき、統計モデルにもよく合致します。この枠組みはまた、系統ラベルが基礎となる遺伝的履歴と矛盾している可能性のある箇所を浮き彫りにし、組換え体や問題のあるゲノムを詳しく調べるようフラグを立てます。データが乏しい場合の過学習や重度に汚染されたサンプルの影響といった課題は残るものの、本研究はパンデミック規模のより信頼できる進化樹を構築することが現実的になったことを示しています。一般読者にとっての結論は、エラーと変異ホットスポットをより適切に扱うことで、病原体がどのように広がり変化するかについての洞察が鋭くなり、将来の流行に対して科学者や保健機関がより迅速かつ確信を持って対応できるようになる、ということです。
引用: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
キーワード: SARS-CoV-2 ゲノミクス, 系統解析手法, シーケンシングエラー, 突然変異率の変動, ゲノム疫学