Clear Sky Science · ja
DiNovoはミラープロテアーゼとディープラーニングにより高カバレッジかつ高信頼度のde novoペプチド配列決定を可能にする
タンパク質をより詳細に見る
タンパク質は細胞を生かす小さな機械ですが、その構成要素を完全に読み取ることは依然として意外に難しい。本論文はDiNovoという新しいソフトウェアシステムを紹介します。これは従来よりもはるかに完全かつ確実にタンパク質断片を「読み取る」手助けをします。巧妙な生化学的手法と最新の人工知能を組み合わせることで、従来法では見落としがちな未知のタンパク質、疾患マーカー、免疫標的などを明らかにする可能性があります。
なぜペプチド断片の読み取りは難しいのか
今日の多くのタンパク質解析は、タンパク質をペプチドと呼ばれる小さな断片に切断し、それらを質量分析計で断片ごとに質量を測る手法に依拠しています。これらの質量情報から、コンピュータが元のペプチド配列を再構築しようとしますが、部分的な手がかりからクロスワードを解くような作業です。既存の手法は通常、ペプチドが既知のタンパク質データベース由来であると仮定します。これは既知のタンパク質には有効ですが、新規や予期せぬものには弱い。いわゆるde novo(デノボ)配列決定はこの制約を避け、データから直接ペプチドを読み取ろうとしますが、断片が欠けやすいことやペプチドがきれいに切断されないことが多く、十分に機能しないことがしばしばあります。
ギャップを埋めるミラー酵素の活用
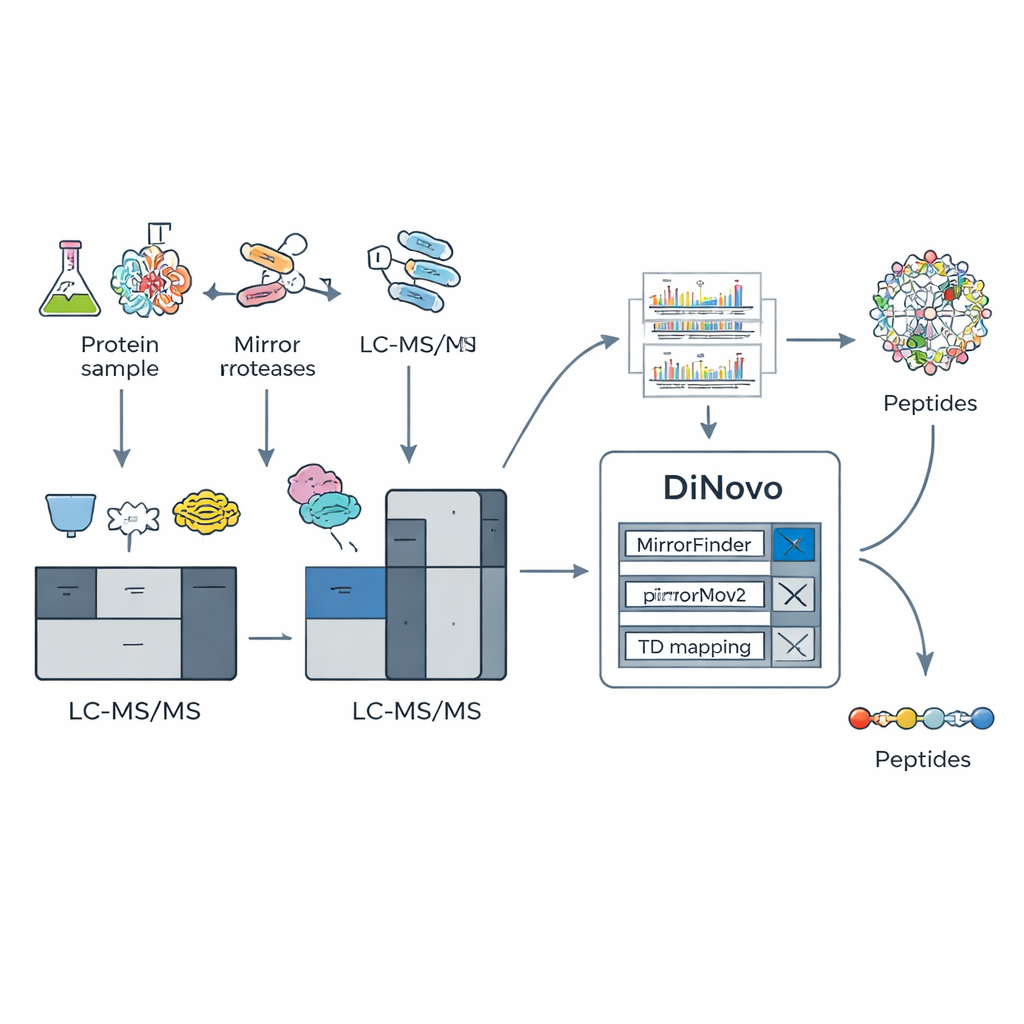
DiNovoの鍵となる発想は「ミラープロテアーゼ」のペアを使うことです。これは同じ種類のアミノ酸の反対側を切断する一対の酵素を指します。たとえば、ある酵素はリジンの直前で切断し、対応する酵素はそのリジンの直後で切断します。こうして得られる二つの関連するペプチドは内部の同じ配列部分を共有しつつ末端が異なります。これらの“ミラー”ペプチドを解析すると、各スペクトルには補完的な断片パターンが現れ、一方のスペクトルで欠けている情報がもう一方に現れることが多いのです。著者らは、このようなミラーペアを組み合わせることで断片カバレッジをほぼ完全に近い水準まで高められることを示しており、実験信号で支持される可能性のある切断位置の約98%が検出されると報告しています。これは単一酵素のみを用いた場合より遥かに高い値です。
ミラーデータ向けに設計されたスマートなソフトウェアパイプライン
この生化学的手法を活用するために、チームはDiNovoをエンドツーエンドのソフトウェアワークフローとして構築しました。まず、細菌や酵母のタンパク質を二組のミラーペア酵素で消化し、得られたペプチドを高分解能質量分析で解析します。DiNovoはまずMirrorFinderというモジュールで、スペクトルのパターンから事前の配列推定を行うことなくどのスペクトルがミラーペアに対応するかを自動で認識します。次に主要なde novoエンジンであるMirrorNovoがこれらのペアスペクトルをディープラーニングで解釈し、バックアップとしてグラフベースのエンジンpNovoM2がより高速なCPUのみのオプションを提供します。これらのツールはピークをアミノ酸配列に変換し、明確なペアを形成しなかった個々のスペクトルも併せて解析して可能な限り情報を引き出します。
既存データベースに依存しない信頼性の測定
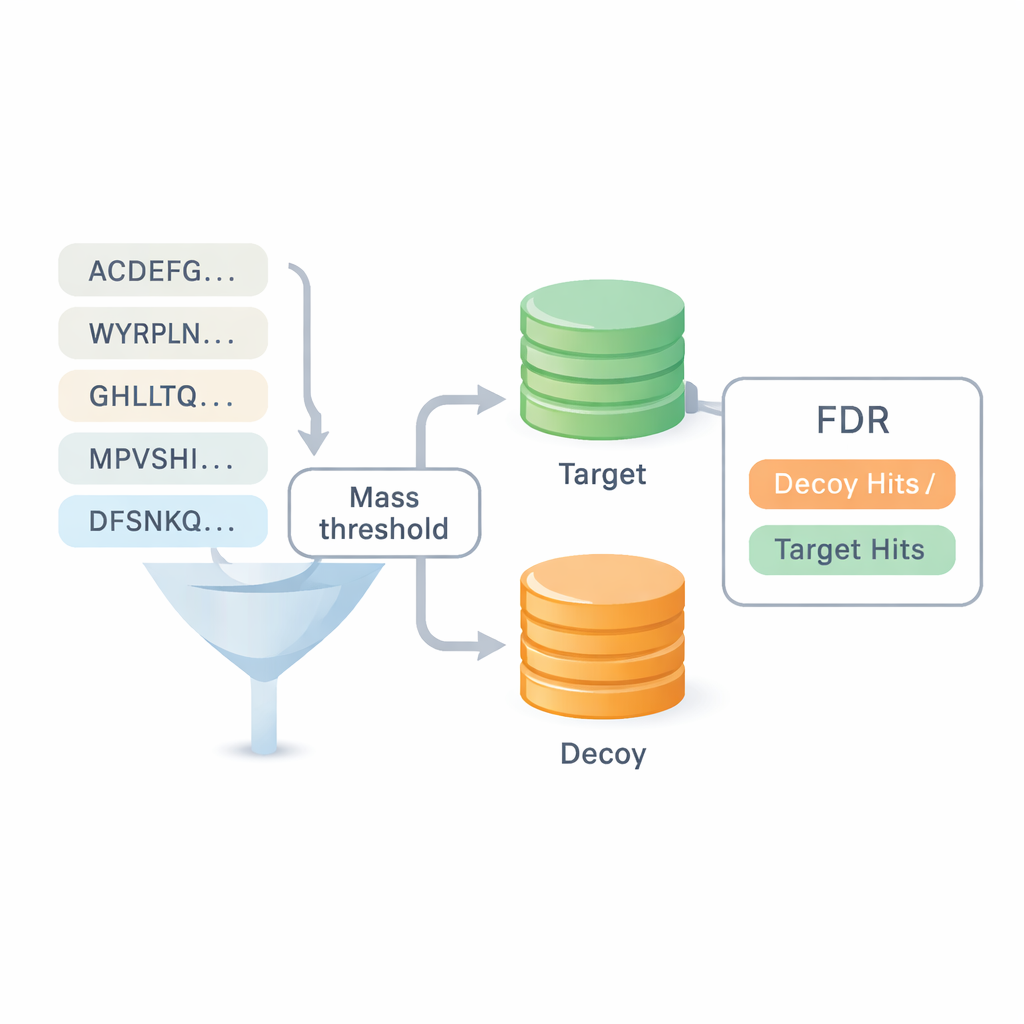
de novo配列決定で最大の課題の一つは、得られた結果をどの程度信頼するかです。既存の多くのベンチマークはデータベース検索の結果を流用しており、両者の境界を曖昧にし、誤りを隠すことがあります。DiNovoはターゲット・デコイマッピングという別の品質検査法を導入します。ここでは、新たに読み取られたペプチドを実在する(ターゲット)配列と人工的にシャッフルした(デコイ)配列を併せたコレクションにマッピングします。ペプチドが実在セットとシャッフルセットのどちらにどれだけ割り当てられるかを比較することで、過去の同定に依存せずに誤認率(false discovery rate)を推定できます。これにより、同一の誤差管理の下でDiNovoを標準的なデータベース検索プログラムと直接比較することが可能になります。
実際にDiNovoがもたらすもの
細菌、酵母、抗体サンプルでの検証では、DiNovoは単一酵素のみを用いる既存の有名なde novoツールよりも一貫して多くのペプチドやアミノ酸を読み取りました。二組のミラーペアを用いることで、古典的なトリプシンのみのセットアップに比べて高信頼度のアミノ酸を2〜3倍多く生成し、同等の誤差レベルでより多くのタンパク質を同定しました。三つの主要なデータベース検索エンジンと直接比較した場合でも、アミノ酸数やタンパク質数は同等であり、同じスペクトルに対する配列の多くは検索エンジンの結果と一致しました。著者らは、この程度のカバレッジと一致度があれば、長く補助的手法と見なされてきたde novo配列決定がデータベース検索と肩を並べ、場合によってはそれを上回る主要な選択肢になり得ると主張しています。
大局的見地:完全で偏りのないタンパク質読み取りに向けて
専門外の方への要点としては、DiNovoは参照データベースに限定されることなく、ペプチド断片を正確に読み取ることを大幅に容易にするということです。支持される配列情報量を二倍〜三倍に増やし、独自の誤り検出機能を提供することで、この手法は未知のタンパク質の発見、微細な変異の追跡、まだ多くの構成成分が不明な複雑な混合物の解析に道を開きます。要するに、ミラープロテアーゼをディープラーニングと厳密な統計手法と組み合わせることで、ノイズの多いスペクトルから健康と疾患の基盤となるタンパク質のより明瞭で信頼できる像を作り出すのにDiNovoは寄与します。
引用: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
キーワード: プロテオミクス, de novoペプチド配列決定, 質量分析, ディープラーニング, ミラープロテアーゼ