Clear Sky Science · ja
亜メソスコピック未満のタンパク質複合体における大規模遷移経路と中間構造の効率的サンプリング
動くタンパク質を観る
私たちの生命を支える多くの分子は、剛直なブロックというよりも絶えず形を変える小さな機械のように振る舞います。これらの運動はエネルギー生産、DNA修復、ウイルスの細胞侵入などの過程を駆動します。クライオ電子顕微鏡のような実験はこうした形のいくつかを凍結したスナップショットとして捉えられるようになりましたが、その間に起こる短い過程は捕らえにくいままです。本稿では、eBDIMS2という新しい計算手法を紹介します。これは、従来は一般的な計算機では扱えなかった巨大な分子機械についても、運動の「欠けたフレーム」を補うことができます。
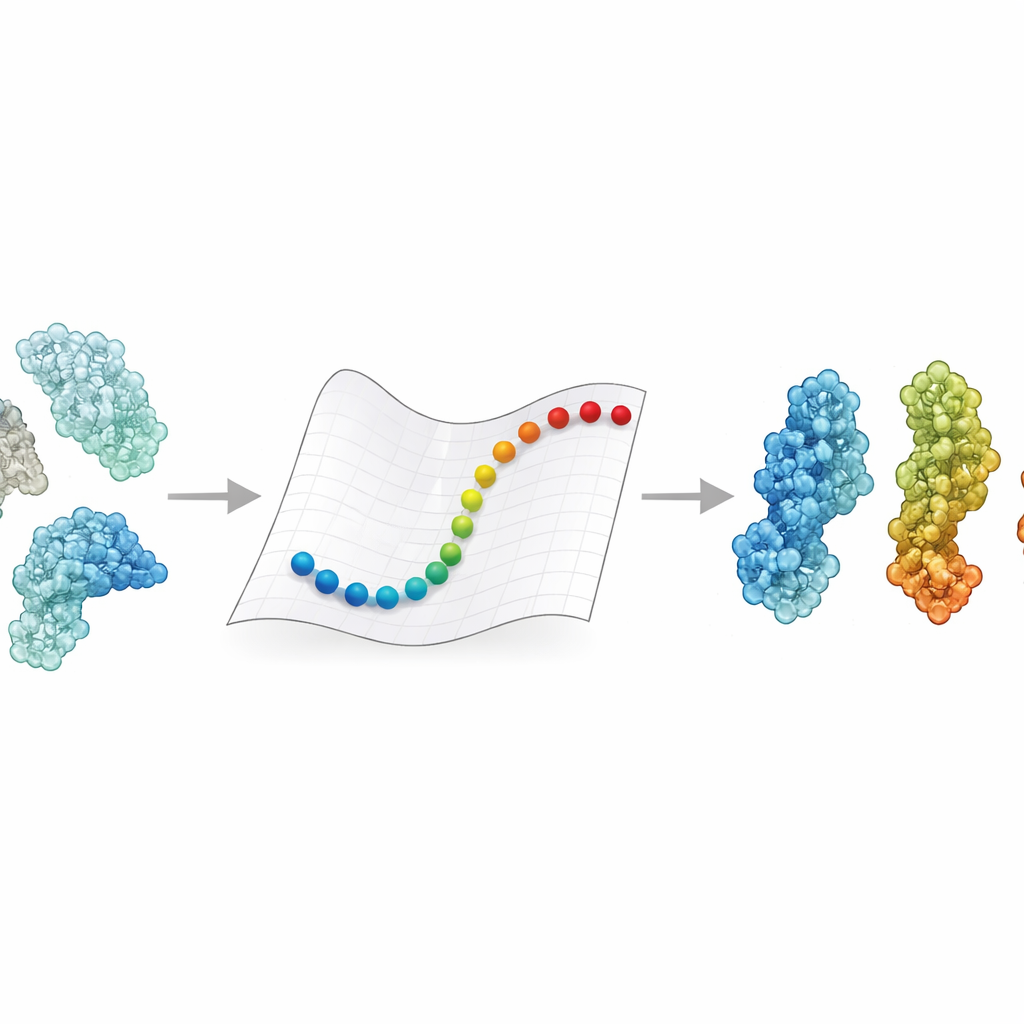
タンパク質の形変化が重要な理由
タンパク質はめったに一つの姿勢に固定されません。電位変化、pH、あるいは結合相手の付着といったシグナルに応じて開閉し、ねじれ、曲がります。これらの変化は酵素が活性化されるか否か、受容体がウイルスを捕捉するか逃すかの違いを生むことがあります。実験は主要な形の詳細なスナップショットを与え、分子動力学シミュレーションは理論上それらを時間的に原子単位で追跡して結びつけられます。しかし、クライオ電子顕微鏡で得られるような数十万〜数百万ダルトン規模の巨大複合体の運動を追うには、通常スーパーコンピュータと数週間単位の計算が必要です。そのため、多くの医療的に重要な巨大分子については、ある状態が別の状態へどのように変化するかがまだ分かっていません。
タンパク質のランドスケープを速く縦断する道
eBDIMS2は、タンパク質の表現と運動の計算方法を単純化することで近道を取ります。すべての原子を追う代わりに、各アミノ酸を弾性ネットワーク内の単一の点として扱い、それらをばねで結びます。これらのばねはタンパク質の異なる部分がどのように一緒に動く傾向があるかを捉えます。次に手法はブラウン運動の数理を用いて、液体中での揺らぎを模した規則に従い、実験的に知られた一つの状態から別の状態へと構造を促します。重要なのは、eBDIMS2が本当に重要な相互作用だけに注目し、距離カットオフと並列計算を使って計算コストを削減する点です。これにより、計算規模はほぼ二乗則からほぼ線形へと改善します。実際には、約二百万ダルトンに迫る巨大なアセンブリの遷移でも、デスクトップ上で数時間で探索できるようになります。
経路を実際のタンパク質と照合する
これらの高速な経路が生物学的に妥当かを確かめるため、著者らは47種類の大型タンパク質とさらに15の複合体から成るアンサンブルを組み、主にクライオ電顕で解かれた数百の構造を集めました。彼らは主成分解析という、各タンパク質が取りうる主要な動き方を抽出する統計手法を用いて、これらの構造を開いた状態、閉じた状態、活性型、非活性型といった立体配座のランドスケープに配置しました。eBDIMS2はこのランドスケープ上で終状態の対を結ぶように求められました。得られた経路は同じ低次元マップに再投影され、実験的に観察された中間体の近傍を滑らかに通るかが明らかにされました。システムの30%以上では、シミュレーション経路が数Å以内で入力として与えられていない中間構造に近づきました。DNA修復酵素DNA-PKcsやコロナウイルスのスパイクタンパク質のような難しいケースでも、粗視化経路はターゲット化分子動力学や高度な強化サンプリングを含むはるかに高コストな原子レベルのシミュレーションとよく重なりました。
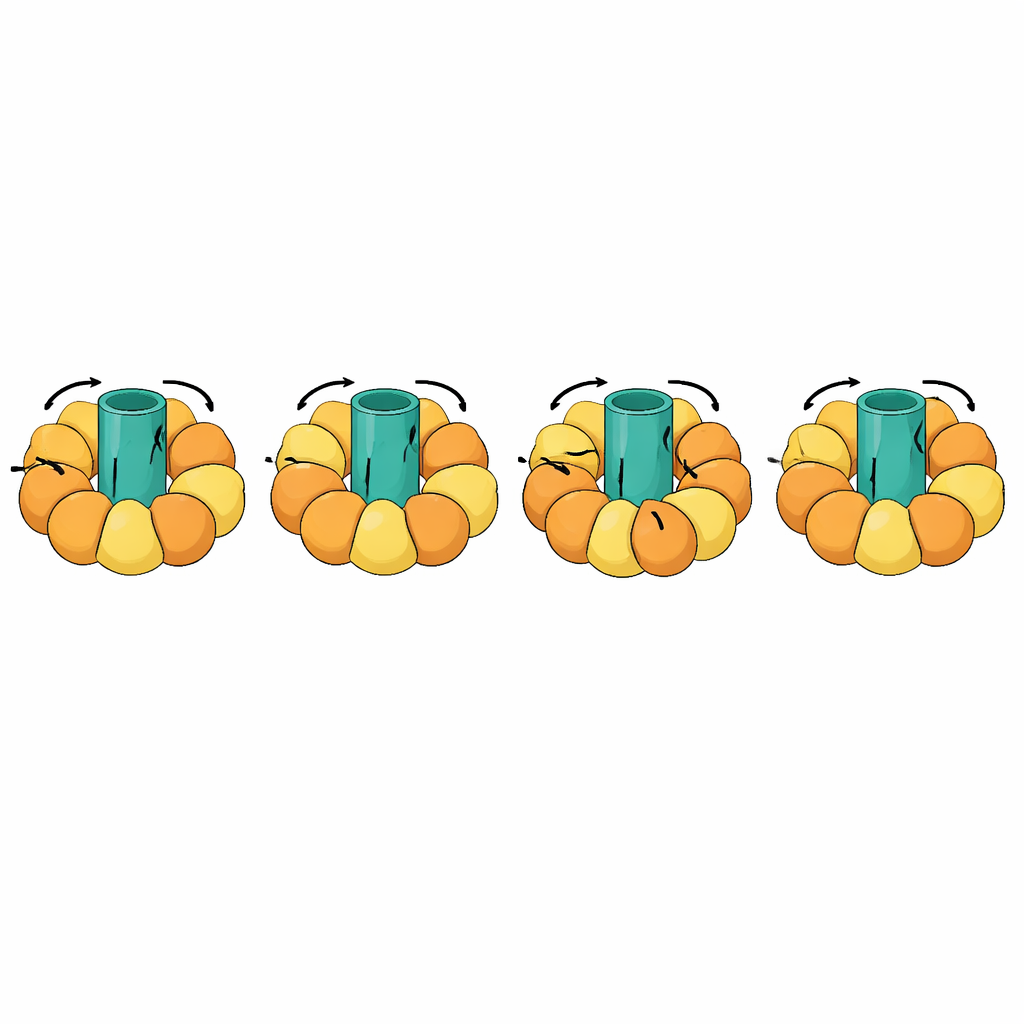
巨大な分子機械の追跡
最も印象的なテストの一つは回転機構を持つATP合成酵素のような機械でした。これらは膜中の回転ロータと周囲のサブユニットの開閉運動を結びつけて細胞のエネルギー通貨を作ります。これらの遷移は極めて複雑で、分子の一部は剛体として回転する一方、他の部分は振付けられた周期でたわむ必要があります。eBDIMS2はそのような準剛体部分や、クライオ電顕でよくみられる欠測セグメントのある不完全な実験モデルに対する特殊な扱いを導入しています。これらの機能により、ATP合成酵素やシャペロン、受容体、ウイルス集合体のような巨大複合体の完全な回転サイクルをシミュレートできます。生成された中間構造は、競合する一部の手法で生じるような深刻な歪みを避け、薬剤設計計算やより長く詳細なシミュレーションに適した原子モデルへと精製できます。
生物学・医学への意義
本研究は、eBDIMS2が従来のシミュレーションでは手の届かなかった系についても、既知のタンパク質形状間の主要な経路を信頼して描けることを示しています。これは詳細な原子レベルのムービーや正確なエネルギーや時間情報を置き換えるものではありませんが、実験で得られた二つの構造を入力として用いるだけで、大規模な分子機械がどのように動く可能性があるかを速くかつ物理に基づいて地図化する手段を提供します。構造データベースにがん、感染症、その他の疾患に関連する大規模タンパク質複合体の複数状態が蓄積されるにつれて、このアプローチは研究者が点をつなぎ、もっともらしい中間状態を提案し、より高解像度の手法や標的を絞った薬剤設計で次に探すべき場所を導くための手軽なツールとなります。
引用: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
キーワード: タンパク質ダイナミクス, 分子シミュレーション, クライオ電子顕微鏡, 立体構造の遷移経路, 粗視化モデリング