Clear Sky Science · ja
単一細胞ATAC-seqデータ解析のためのコンセンサスピークによって定義された汎用参照
なぜDNAの「開いた扉」を地図化することが重要か
体内のほぼすべての細胞は同じDNAを持ちながら、神経細胞、血液細胞、腫瘍細胞は非常に異なる振る舞いをします。その大きな理由の一つは、ある時点でどの領域のDNAが露出して「開いている」かが異なるためです。最近の単一細胞技術ではゲノム全体のこの開放状態を測定できますが、これまで実験や研究室間で結果を比較するための共通の参照地図、いわば標準アトラスが欠けていました。本研究はcPeaksと呼ばれるそのような地図を構築し、細胞型、発生、癌の解析をどのように鋭くするかを示します。
多数の実験を一つの共有地図に統合する
著者らはまず、40以上のヒト臓器にまたがる開放クロマチン(DNAのアクセス可能な部分)を測定した624件の高品質実験を収集しました。各実験ではコンピュータプログラムが特に露出した「ピーク」を既にマークしていました。各データセットを個別に扱うのではなく、チームはこれらすべてのピークリストをゲノム上に慎重に重ね合わせ、重複する領域を統合しました。次に、統合領域内の微小な位置ごとに、どれだけ頻繁に開放と呼ばれたかを調べ、それぞれの領域を出現頻度を反映する特徴的な形状に変換しました。統合領域に近接した複数の開放部位が含まれている場合は、それを複数の単純な単位に分割しました。これらの単位は合計約140万に達し、観測されたコンセンサスピーク(cPeaks)として、ヒトのクロマチン可及性の候補参照カタログになりました。 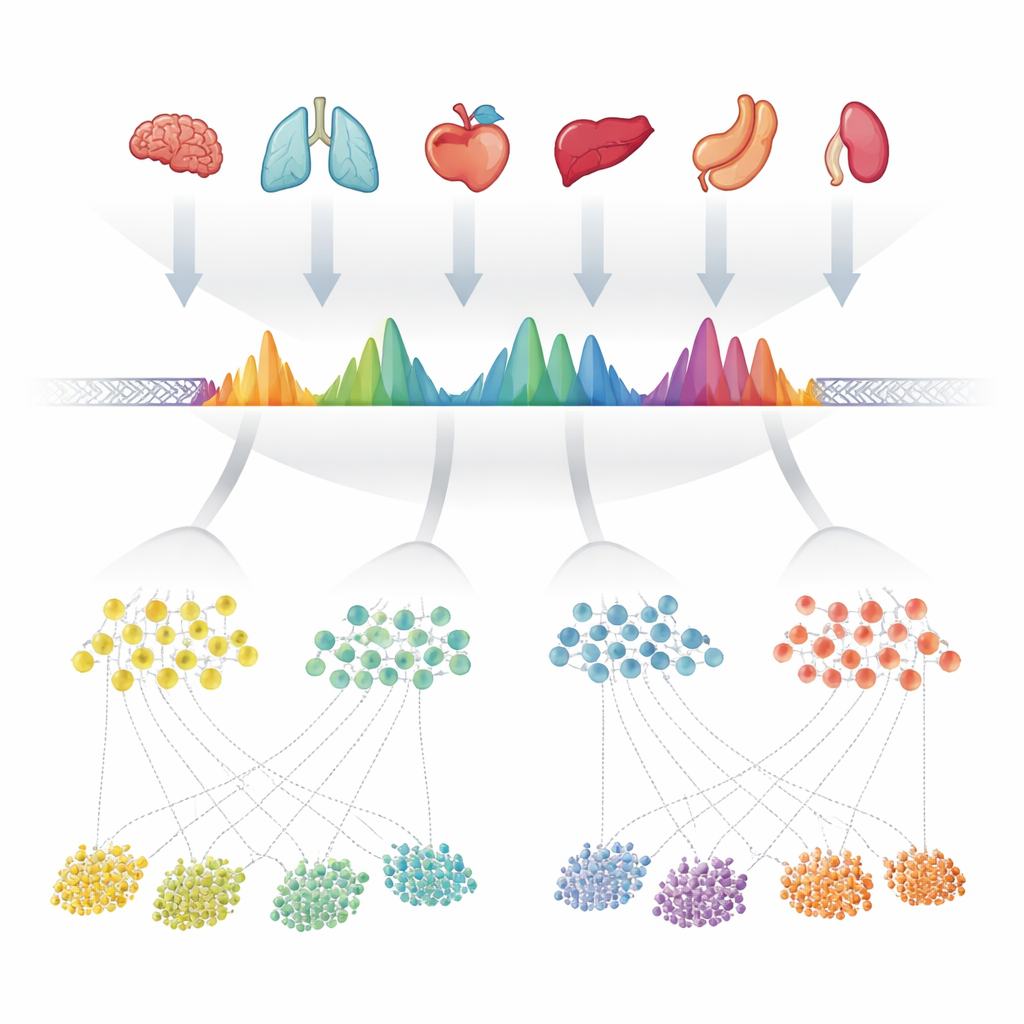
組織や技術を超えて安定した指紋
有用な参照であるためには、cPeaksは特定のサンプルやソフトウェアの癖ではなく、ゲノムの真正で再現性のある特徴を表していなければなりません。著者らはこれを検証するため、統合領域を血液サンプルのみ、固形組織のみ、別の公開データベースのみ、さらには開放DNAを検出する異なる実験手法ごとに再作成しました。いずれの場合も、同じゲノム位置は驚くほど類似したピーク形状を示し、検討したほとんどの単一細胞データセットは自身のピークの90%以上をcPeakカタログと重ね合わせました。多くの臓器からのリードはcPeak中心の周辺に正確に集積し、これらの領域がクロマチンの開放位置を確実に捉えていることを示しました。関連技術に基づく従来の参照セットと比べて、cPeaksはATAC-seq実験で検出される可及領域をより広く覆い、各データセットで新たに定義されたピークとほぼ同等の信号を捉えていました—固定かつ再利用可能でありながらです。
欠けた領域を見つけるためにニューラルネットワークを教える
既存のサンプルが数百件あっても、すべての細胞型をカバーするわけではありません。未観測の領域に地図を延長するために、チームは深層学習に目を向けました。彼らはDNA配列を入力とした1次元畳み込みニューラルネットワークを訓練しました:観測されたcPeaks内にある例を陽性、ランダムに選んだ背景領域を陰性として与えました。モデルは高い精度でこれらを識別することを学び、cPeaksが認識可能な配列パターンを持つことを示唆しました。研究者らがある組織特異的なピークを意図的に隠した場合でも、ネットワークは配列情報だけでそれらを回復し、稀な組織特異的サイトも含めて復元しました。その後、ゲノムの残り領域に沿って小さなウィンドウをスライドさせ、各セグメントをスコアリングして約28万件の高スコア新規領域を予測cPeaksとしてカタログに追加し、特に元データで過小表現されていた組織のカバレッジを改善しました。
開放領域を遺伝子、細胞型、稀な細胞に結びつける
より豊富な参照を得て、著者らはこれらの領域の機能を問い直しました。多くのcPeaksは遺伝子の開始部位や終了部位の近傍にあり、プロモーターやエンハンサー、CTCFのような構造タンパク質の結合部位など既知の調節要素と重複していました。ごく一部はほぼすべてのデータセットでアクセス可能であり、これらのより長い「ハウスキーピング」cPeaksは基本的な細胞維持に必要な遺伝子のコアプロモーター領域に位置する傾向がありました。チームはまた、サンプル間で境界がどれほど鋭く一貫しているかに基づいてcPeaksを分類しました。これは周辺のDNAがヌクレオソームとしてどれほど正確に配置されているかを反映します。境界が鋭く定義される領域は、クロマチンを再編成し発生を駆動することが知られた特定の転写因子ファミリーに富んでいました。cPeaksを特徴セットとして複数の単一細胞データセットを解析すると、細胞型ラベリングの精度が向上し、特に稀な細胞型や以前のピーク集合や単純なゲノムグリッドではしばしばぼやけていた微妙なサブタイプの同定に有用でした。
共通言語で発生と癌を追跡する
標準参照の威力は非常に異なる生物学的文脈を比較したときに明らかになります。著者らはcPeaksを用いて、発生中のヒト網膜、胎児および成人組織の大規模アトラス、いくつかの癌からの単一細胞データを再解析しました。彼らは発生軌跡を再構築でき、鋭く境界が定まった「位置決めの良い」cPeaksの割合が過渡期には上昇し、細胞が安定した同定に落ち着くと減少する傾向があることを確認しました。類似のパターンは腫瘍段階全体にも現れ、中間段階の癌はこれらの構造化された領域の割合が高く、激しい調節再編成が起きていることを示唆しました。ある卵巣腫瘍では、cPeaksが異なるDNAコピー数変化を持つ二つの明確な癌細胞サブクローンを明らかにするのに役立ち、参照が疾患の隠れた複雑性を暴く例を示しました。
今後のゲノム研究にとっての意義
非専門家向けに言えば、cPeaksは多くのヒト細胞型にまたがってゲノムが物理的に開いて活性化している可能性が高い座標を標準化したセットと考えられます。新しい単一細胞クロマチン実験をこの共有地図に合わせることで、研究者は研究間で結果を比較しやすくなり、稀なまたは過渡的な細胞状態を見つけやすくなり、遺伝子調節の大規模なモデルを構築し始めることができます—標準化された遺伝子カタログが単一細胞RNAアトラスの発展を可能にしたのと同様に。現在のcPeakカタログは新たなデータの到着とともに成長していく初稿ですが、すでにクロマチン可及性を記述する共通言語を提供しており、DNAのパッケージングが発生、健康、疾患をどのように導くかを統一的に理解することに一歩近づけます。 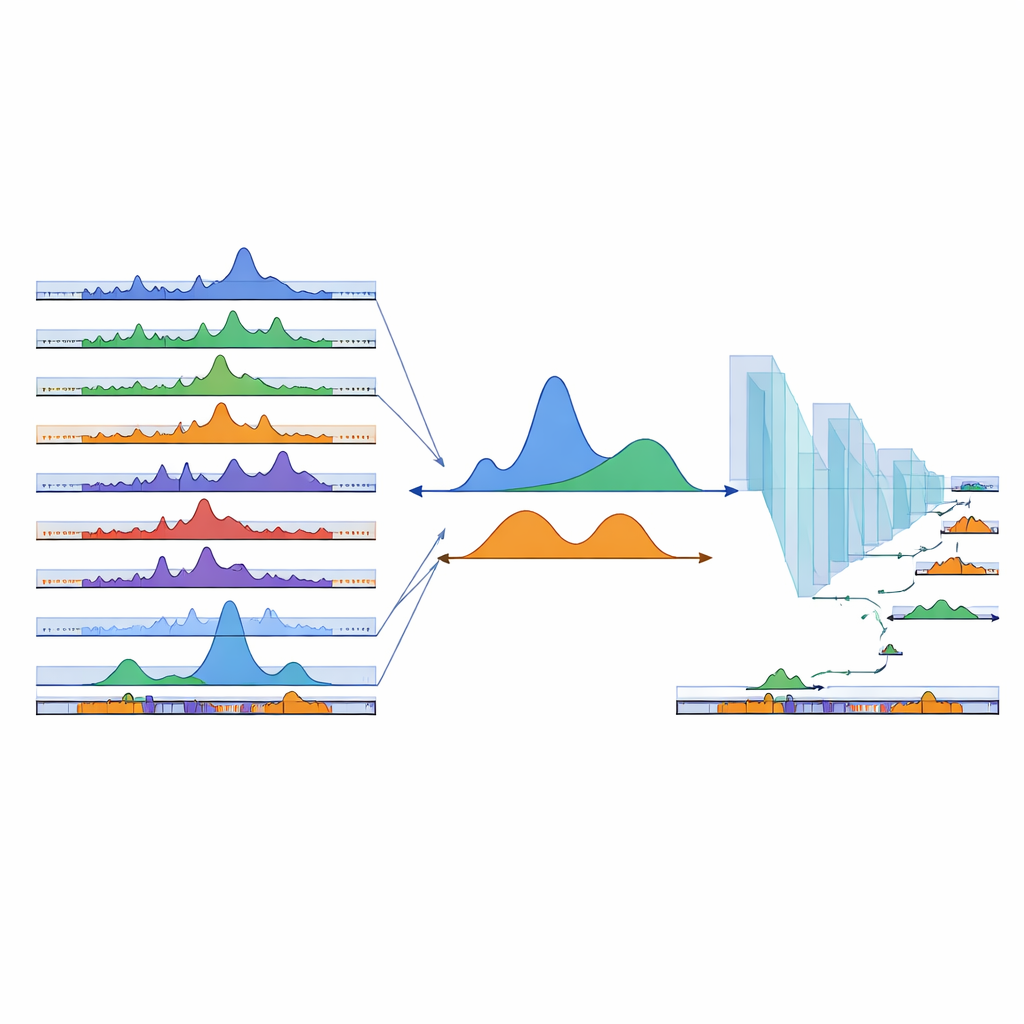
引用: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
キーワード: クロマチン可及性, 単一細胞ATAC-seq, コンセンサスピーク, 遺伝子調節, 深層学習ゲノミクス